

Home
Studies
& Data Analysis
Methods
Microscope studies
Flagella experiment
Laboratory math
Blood fractionation
Gel electrophoresis
Protein gel analysis
Mitochondria
Concepts/ theory
Keeping a lab notebook
Writing research papers
Dimensions & units
Using figures (graphs)
Examples of graphs
Experimental error
Representing error
Applying statistics
Principles of microscopy
Solutions & dilutions
Protein assays
Spectrophotometry
Fractionation & centrifugation
Radioisotopes and detection
Guide to the study
Lab part 1
Lab part 2
Lab part 3
Selected methods
Preparing Protein Samples for Electrophoresis
A polypeptide is a macromolecule consisting of a nonbranching sequence of amino acids, each connected to the next by a single peptide bond. A protein consists of one or more polypeptides and/or additonal types of molecules, held together by any of a number of molecular interactions often including covalent bonds. Such interactions result in several levels of organization, which we call primary, secondary, tertiary, and quaternary structures. Intact proteins are notoriously difficult to separate reproducibly. Patterns of bands vary depending on temperature, buffer, variations in pH, quality of a preparation, etc. To characterize a type of preparation and obtain predictable results we try to take proteins apart so that what we have left is primary structure only. Sample preparation sometimes falls short of that ideal, which you will discover as you analyze your results.
The amino acid sequence of a polypeptide is called its primary structure. Interaction of soluble proteins with water leads to hydrogen bonding, which is partially responsible for the secondary structure of proteins. Secondary structure refers to the local structure of a polypeptide chain, including helices, pleated sheets, and turns. A functional protein has a three dimensional stucture resulting from hydrogen bonding, hydrophobic amino acids, Van der Waal's forces, and disulfide bonding. Three dimensional structure of a protein is called its tertiary structure.
Quaternary structure refers to the interaction of individual polypeptide chains with other molecules to form functional proteins. Although some proteins do consist of single polypeptides, many consist of two or more polypeptides linked by covalent bonds and/or noncovalent forces. In fact, many native (functional) proteins include nonprotein components such as the heme group of hemoglobin and the carbohydrate groups on many membrane-associated proteins.
Sample denaturation
Various sample buffers have been used for SDS-PAGE but all use the same principles to denature samples. We obtain good denaturation by preparing a sample to a final concentration of 2 mg/ml protein with 1% SDS, 10% glycerol, 10 mM Tris-Cl, pH 6.8, 1 mM ethylene diamine tetraacetic acid (EDTA), a reducing agent such as dithiothreitol (DTT) or 2-mercaptoethanol, and a pinch of bromophenol blue to serve as a tracking dye (~0.05 mg/ml).
We prepare a 2x concentrate of sample buffer consisting of 2% SDS, 20% glycerol, 20 mM Tris-Cl, pH 6.8, 2 mM ethylene diamine tetraacetic acid (EDTA), 160 mM dithiothreitol (DTT), and 0.1 mg/ml bromphenol blue dye. I prefer DTT to 2-mercaptoethanol because the latter has a much stronger unpleasant odor and it doesn't denature our blood fractions very well. Part of the problem is that our water baths don't reach the boiling point, and boiling may be necessary with 2-mercaptoethanol. We prepare all of our unknowns to the same concentration then mix 1 volume prepared sample to 1 volume 2x buffer.
So, what do the various components do? EDTA is a preservative that chelates divalent cations, which reduces the activity of proteolytic enzymes that require calcium and magnesium ions as cofactors. The tris acts as a buffer, which is very important since the stacking process in discontinuous electrophoresis requires a specific pH. Glycerol makes the sample more dense than the sample buffer, so the sample will remain in the bottom of a well rather than float out. The dye allows the investigator to track the progress of the electrophoresis.
SDS, DTT, and heat are responsible for the actual denaturation of the sample. SDS breaks up the two- and three-dimensional structure of the proteins by adding negative charge to the amino acids. Since like charges repel, the proteins are more-or-less straightened out, immediately rendering them functionless. Some quaternary structure may remain due to disulfide bonding (covalent) and due to covalent and noncovalent linkages to other types molecules. By the way, another name for SDS is lauryl sulfate. Your shampoo may contain lauryl sulfate - now doesn't that inspire confidence in the product?
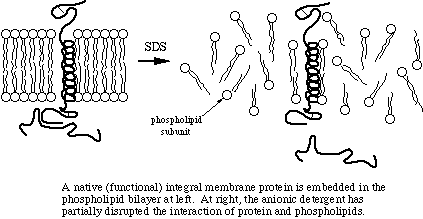
Many proteins have significant hydrophobic properties and may be tighly associated with other molecules, such as lipids, through hydrophobic interaction. Heating the samples to at least 60 degrees C shakes up the molecules, allowing SDS to bind in the hydrophobic regions and complete the denaturation.
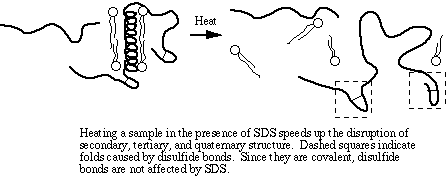
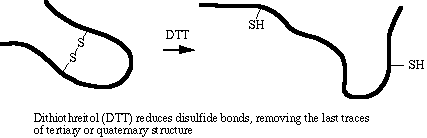
The amino acid cysteine contains a sulfhydryl (-SH) group that spontaneously forms a disulfide bond (-S-S-) with another sulfhydryl group under normal intracellular conditions. Disulfide bonding is covalent and is not disrupted by SDS. DTT is a strong reducing agent. Its specific role in sample denaturation is to remove the last bit of tertiary and quaternary structure by reducing disulfide bonds.
Most sample buffers do not remove covalently attached carbohydrate or phosphate groups, and some associations with other types macromolecules are difficult to disrupt. Polypeptides contain varying amounts of basic and acidic amino acids that add charge to the molecules, and individual amino acids vary in molecular weight although they may bind SDS with the same affinity. Therefore, charge to mass ratio and the relative mobility of many proteins is affected by factors other than strictly the molecular weight. SDS-PAGE is very effective in providing reproducible results, but don't count on precise values for MW determination.
Amounts to load
Polyacrylamide has a limited capacity for protein. Overloading results in precipitation and aggregation of proteins, producing streaks and smears. Underloading results in complete disappointment, as one may detect only the most abundant of polypeptides, if that. The objectives of sample preparation are to put the proteins into a denaturing buffer, rendering them suitable for electrophoresis, and to adjust the concentrations of sample so that an appropriate amount of protein can be loaded onto a gel.
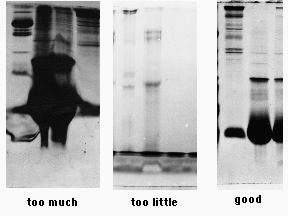
We get the best results if we load 10 µl of a 2 mg/ml final concentration of denatured protein per sample well. While some of the more concentrated proteins will be overloaded, we will detect bands that represent the less common ones. A typical mini-gel well holds 10 µl easily, and perhaps 20 µl or more if the well dividers are in good shape.
We will dilute all samples to a predetermined concentration and volume before mixing with the denaturing buffer. Efficient laboratory personnel divide responsibilities, so that while gels are polymerizing they are preparing the samples themselves, to volumes that are at least double the minimum needed to fill the sample wells. Such people start their work prepared with calculations of the volumes of sample, water, and 2x concentrated sample buffer they need in order to prepare each of their samples for electrophoresis.
To completely denature the samples we heat them in a steaming water bath for at least 10 minutes. Standards for molecular weight determination are prepared the same way. They are expensive, and although the suppliers give instructions for mixing, it is usually necessary to test them and to make adjustments before relying on them for internal calibration of an important gel. A "dirty" sample (containing a lot of particulate matter) should be centrifuged just before loading. However, samples containing soluble proteins only and samples from a typical blood fractionation are so "clean," that centrifugation is not necessary.
Notes on sample preparation
- In a materials and methods section an investigator reports the general procedure used for sample preparation. It is amateurish to report the volume calculations for each and every sample. Such information has no relevance for other investigators. Your reviewers and/or editor would insist on deleting such unnecessary information.
- A proper amount of protein to load depends on the distribution of individual proteins in the sample. If the sample consists of a single, nearly pure polypeptide, 10 micrograms would give a huge blob. A rule of thumb for mini-slab gels is to load about 0.5 microgram protein per expected band. Since complex mixtures contain proteins of widely varying concentrations, there is no ideal single amount to load.
- Heating simply speeds up the process of denaturation by increasing molecular motion. It isn't necessary for some samples, but is necessary for membrane samples.
- Heating to the boiling point can cause aggregation of proteins, defeating the purpose of SDS-PAGE. Insufficient heating can leave some proteins incompletely denatured. It may require trial and error to achieve the best results.
- Once denatured, the samples can sit on a benchtop at room temp until it is time to load them. If they are to be saved for another day, they should be frozen.
- If samples are heated without first mixing with sample buffer, they will indeed be denatured, but not in the intended manner. Think of what happens when you boil an egg.
Visitors: to ensure that your message is not mistaken for SPAM, please include the acronym "Bios211" in the subject line of e-mail communications
Created by David R. Caprette (caprette@rice.edu), Rice University 19 Aug 96
Updated 30 Jul 12