

Home
Studies
& Data Analysis
Methods
Microscope studies
Flagella experiment
Laboratory math
Blood fractionation
Gel electrophoresis
Protein gel analysis
Mitochondria
Concepts/ theory
Keeping a lab notebook
Writing research papers
Dimensions & units
Using figures (graphs)
Examples of graphs
Experimental error
Representing error
Applying statistics
Principles of microscopy
Solutions & dilutions
Protein assays
Spectrophotometry
Fractionation & centrifugation
Radioisotopes and detection
Guide to the study
Lab part 1
Lab part 2
Lab part 3
Selected methods
Analysis of Protein Gels (SDS-PAGE)
The resources on protein gel analysis focus on "routine" gels that are use to separate polypeptides from samples containing a mix of proteins. Such gels are most often stained with Coomassie blue dye, although the principles described here also apply to gels stained by other means. Before starting an analysis one's goals should be defined. Analysis of every band resolved on a gel, no matter how faint, can take a lot of time. Here are some observations that one might make using a Coomassie stained gel.
- Estimated molecular masses and relative abundance of unknown polypeptides in a complex mixture
- Patterns of bands that suggest presence of isoenzymes or specific complex proteins
- Effectiveness of a separation procedure during cell/tissue fractionation
- Effectiveness of a procedure to purify specific organelles, proteins, or polypeptides
- Condition of a preparation such as extent to which proteins have degraded
- Similarity of one preparation to another
- Changes in gene expression during developmental stages or resulting from experimental intervention
Semi-quantitative analysis of a gel begins with a cursory examination to see if the results make sense. Often an investigator repeatedly runs samples that give reproducible patterns, for example. A change in the pattern could indicate that something was wrong. The following steps describe how one might calibrate and interpret a gel on which was run a series of aliquots from a fractionation procedure.
- Identify top/bottom and left/right
- Identify which lane corresponds to which sample
- Identify the lane or lanes to be analyzed
- Assess the success of the fractionation – do fractions overlap, that is, "share" the same polypeptide band(s)?
- Calibrate the gel using standards of known molecular mass (set up a standard curve if necessary)
- Select polypeptide bands in the lane(s) of interest to be analyzed and identify them by some generic label (e.g., a, b, c,... or 1, 2, 3,...)
- Estimate molecular mass or relative molecular mass for each band of interest
- Note differences in intensity of staining that reflect relative abundance of individual polypeptides
- Note unusual patterns that might indicate isoenzymes, incomplete denaturation, degradation, etc.
- Note qualitative differences among bands that suggest presence of hydrophobic regions and/or covalent bonding to non-protein substituents
- Use the available information to characterize the unknowns
This method does have limitations. For example, identification of a band on a protein gel is not considered positive proof of identity. A great many different polypeptides have very similar molecular masses. One band may mask the presence of more than one polypeptide. Incomplete denaturation, unusual amino acid sequences, and/or presence of non-protein residues can affect mobility, resulting in considerable error estimating molecular mass. We refer to estimates from gels as "apparent molecular mass" for that reason. A unique band may be nothing more than a product of degradation of a heavier polypeptide or an aggregate of two or more lighter ones.
Orientation
Obviously one must have a clear record of how the sample wells were loaded. To maintain left/right orientation one can load the standards to one side of the gel and/or mark the bottom left or right of the gel by taking a piece out just before staining.
Bromphenol blue tracking dye diffuses out of acrylamide gels during staining and destaining. If a "dye front" remains it is there because of smaller Coomassie stained polypeptides that were not resolved. When all proteins in a mix are resolved, the original position of the tracking dye is lost following staining/destaining. If measurement of relative mobility is critical, the position of the tracking dye should be marked before staining the gel. Some investigators push a thin wire into the gel edges to mark the dye front. When standards of known molecular mass are run with the unknowns, estimates of molecular mass can be made using some arbitrary reference point such as the bottom of the gel for calculating relative mobility.
Critique your gel
Overloading, underloading, distortion of lanes, non-reproducible patterns, smears, streaks, etc. can limit the useful information that you can obtain. If such issues prevent you from extracting the information you need, perhaps the "hall of shame" can help you match a cause to your symptoms.
Calibrate the gel
The stacking gel is of no use to the analysis and it can be removed. Top of the gel refers to the top of the separating gel, that is, the point at which different polypeptides began to separate. A mix of protein standards usually consists of five to eight individual polypeptides that produce a prominent "ladder." Standards are identified from the top down. Depending on %T one or more of the lowest mass standards may not resolve.
To prepare a standard curve for molecular mass one estimates a relative mobility for each standard and plots a standard curve of molecular mass versus relative mobility on semi-log paper or log molecular mass versus relative mobiltiy on conventional graph paper. Relative mobility is determined by measuring the distance from the top of the gel to the middle of the dye front or arbitrary reference point, measuring the distance from the top of the gel to the middle of the band, and dividing the second measurement by the first. This is the Rf, which is always between 0 and 1.
Note that the relative mobility of a given protein depends on gel concentration. Any single gel has an upper and lower limit to its useful range for estimating molecular mass.
Estimate apparent molecular mass for unknowns
Relative mobility should be calculated for each band of interest and the standard curve used to estimate apparent molecular mass. Because the relationship between mass and Rf is logarithmic, one should interpolate the standard curve data rather than use a trendline that may miss some of the data points. It is especially important to avoid extrapolating the standard curve, since even the logarithmic relationship begins to break down in the top 20% or so of a gel. One can report the mass of an unknown to exceed that of the highest mass standard, but cannot estimate a molecular mass for an unknown with Rf smaller than that of any of the standards. For example, if the Rf for the myosin standard (205 kDa) was 0.18 and the Rf for unknown 1 was 0.15, one reports that unknown 1 had apparent molecular mass > 205 kDa.
Consider resolution in the appropriate region of a gel and thickness of the band of interest when determining significant figures with which to report a mass estimate. For example, suppose the distance between 97,000 and 116,000 kDa standards is 0.5 cm and a band between them is 1 mm thick. You have resolution to the nearest 4,000 Daltons. An estimate of, say, 110 kDa should probably be written as 110 ± 4 kDa.
Qualitative analysis
In addition to looking for a reproducible pattern one should be on the lookout for unusal associations of bands. For example, it is unlikely that two unrelated polypeptide bands of very similar staining intensity would show up close together on a gel. We refer to such an association as a "doublet." A doublet often indicates presence of two polypeptides that share the same amino acid sequence for the most part, although one may have extra residues and/or they may differ in a few key positions. In nature such polypeptides typically form multiple-subunit proteins or are part of a "family" of isoenzymes that perform similar functions but are each specialized for a specific tissue and/or purpose.

Polypepetides from cytoplasmic proteins or that are membrane-associated but extrinsic to a membrane tend to form well resolved narrow bands with sharp edges. Polypeptides that form intrinsic proteins bear sequences of hydrophobic side chains that may or may not denature completely. They often form bands that are broader with less distinct edges, indicating that the individual molecules ran as if they represented a distribution of molecular masses. Very large bands that are otherwise distinctly formed suggest either overloading of protein in a well or presence of a predominant polypeptide in a sample.
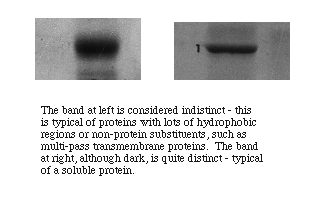
Characterize the membrane-associated proteins
SDS-PAGE of purified organelles such as plasma membranes, ribosomes, endoplasmic reticulum, etc. usually gives patterns that are dominated by one or a few major protein bands. For example, on a 7 to 8% gel, erythrocyte membranes yield a pattern that is dominated by: the heavy (>200 kDa) spectrin bands, which form a doublet at the top; the wide anion exchanger protein band (band 3) in the range of 100 kDa or so; and the actin band at around 43 kDa. An investigator should look for the pattern, double check that the apparent molecular weights are in the right ball park, and use the basic pattern as a framework for identification of less dense bands.
Previously published work or even textbook articles might present gel patterns to which to compare your own results. If an expected pattern is not apparent, there may have been a problem with the fractionation, sample preparation, or the gel itself. Degradation of proteins or failure to completely denature a sample are common reasons for such 'strange' results.
Bands that consistently show up following electrophoresis of a particular sample are very likely representative of the polypeptides that characterize the sample. On the other hand, bands that occasionally appear and/or are rather faint may represent important polypeptides or they may represent something else. Large proteins often tend to degrade. When a large protein is cleaved, two smaller products result, and each product resolves into its own band. If you see a pattern among samples in which the disappearance of a heavy band is correlated with the appearance of one or more smaller bands, you may have degradation.
Bands containing two or more polypeptides may result from the presence of polypeptides that normally form noncovalent associations with each other or with other polypeptide types. Sometimes such associations persist, especially if the polypeptide is present in high relative concentration. For example, hemoglobin in native form consists of four monomers each with molecular mass of about 15 kDa (mass varies among species), associated with a heme group. Samples containing large amounts of hemoglobin may produce as many as four bands corresponding to the monomer, dimer, trimer, or tetramer of hemoglobin.
Visitors: to ensure that your message is not mistaken for SPAM, please include the acronym "Bios211" in the subject line of e-mail communications
Created by David R. Caprette (caprette@rice.edu), Rice University 16 Aug 96
Updated 15 May 05