2019 Abstracts.
134. P. Yu, Z. Wang, M. Marcos-Hernandez, P. Zuo, D. Zhang, C. Powell, A.Y. Pan, D. Villagrán, M.S. Wong, and P.J.J. Alvarez, "Bottom-up biofilm eradication using bacteriophage-loaded magnetic nanocomposites: a computational and experimental study" Environ. Sci.: Nano 6, 3539-3550 (2019) DOI: 10.1039/C9EN00827F (ES Nano Best Paper Collection)
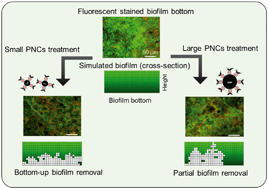
Biofilms cause a variety of pervasive problems in water treatment, distribution and reuse systems that are difficult to mitigate due to their resistance to disinfectants. We used magnetic phage-nanocomposite conjugates (PNCs) to target bacteria in biofilm inner layers for bottom-up eradication. Polyvalent Podoviridae phages PEB1 (54 nm) or PEB2 (86 nm) were covalently conjugated (via amide bonds) with magnetic colloidal nanoparticle clusters (CNCs) of different sizes (150, 250 or 500 nm). Smaller CNCs with higher density of amino groups loaded phages more efficiently than the largest CNCs (e.g., for PEB1, 60 ± 4, 62 ± 5, and 47 ± 4 phages were loaded per μm2). Smaller PNCs dispersed phages more evenly throughout the biofilm bottom, significantly disrupting the biofilm bottom layer and detaching the biofilm within 6 h. The biofilm removal efficiency was 98.3 ± 1.4% for dual species biofilm (i.e., Escherichia coli and Pseudomonas aeruginosa) and 92.2 ± 3.1% for multi-species biofilm (i.e., E. coli, P. aeruginosa, and non-hosts Bacillus subtilis and Shewanella oneidensis). Large PNCs caused higher physical disruption but lower corresponding removal efficiencies (i.e., 80.2 ± 3.4% for dual species biofilm and 67.6 ± 3.8% for multi-species biofilm) due to lower horizontal diffusion at the bottom of the biofilm. A semi-empirical numerical model corroborated the higher biofilm removal efficiency with smaller PNCs and inferred that PNC size influences the mode of phage propagation: Small PNCs facilitate biofilm bottom clearance with significant horizontal dispersion while large PNCs mainly enhance vertical propagation. Overall, this study demonstrates the importance of size control to enhance the biofilm eradication capability of PNCs as an alternative or complementary biofilm control strategy.
133. P. Dias da Silva, S.K. Samaniego Andrade, K. Zygourakis, and M.S. Wong, "Adsorptive Desulfurization of Liquid Fuels at Elevated Temperatures Using Metal Exchanged Zeolite Y" Ind. Eng. Chem. Res. 58, 19623-19632 (2019) DOI: 10.1021/acs.iecr.9b03203
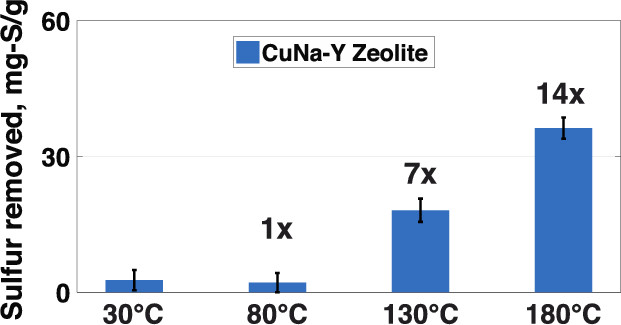
It has long been known that the ability of adsorptive methods to remove sulfur from transportation fuels at room temperature is limited by the competition of aromatic hydrocarbons and organosulfur compounds for the active sites of the adsorbent. In an effort to overcome these limitations, we studied adsorptive desulfurization at temperatures substantially higher than those considered by previous investigators. Na-Y and Cu-exchanged Na-Y (CuNa-Y) zeolites were used to remove sulfur from model fuels and a JP-8 fuel at temperatures up to 180 °C. Batch desulfurization and temperature-programmed desorption experiments with model fuels containing 3-methyl-benzothiophene (3-MBT) and dodecane showed that 3-MBT removal was strongly dependent on treatment temperature and involved weak physisorption bonds with Na, weak interactions with Cu sites at 30 or 80 °C, and strong chemisorption (S–Cu bonding) at 130 or 180 °C. Overcoming the competition that favors the adsorption of aromatics at low temperatures, the formation of strong S–Cu bonds at high temperatures shifted the balance and allowed significant 3-MBT removal even at high toluene concentrations. Desulfurization tests with a JP-8 fuel containing 2230 parts per million weight (ppmw) of total sulfur revealed that elevated temperatures dramatically improved the efficacy of the CuNa-Y zeolite, increasing its sulfur-removal capacity from 2.6 mg of S per gram of adsorbent at 30 °C to 36 mg of S per gram of adsorbent at 180 °C. Sequential desulfurization experiments showed that the total JP-8 sulfur content can be lowered by 95% after four treatments and that CuNa-Y removed the least refractory organosulfur compounds first. Our results suggest that ultradeep desulfurization of JP-8 fuel is achievable using zeolitic adsorbents at elevated temperatures.
132. K.N. Heck, Y. Wang, G. Wu, A. Tsia, D.T. Adamson, and M.S. Wong, "Effectiveness of metal oxide catalysts for the degradation of 1,4-dioxane" RSC Advances 9, 27042 - 27049 (2019)DOI: 10.1039/C9RA05007H
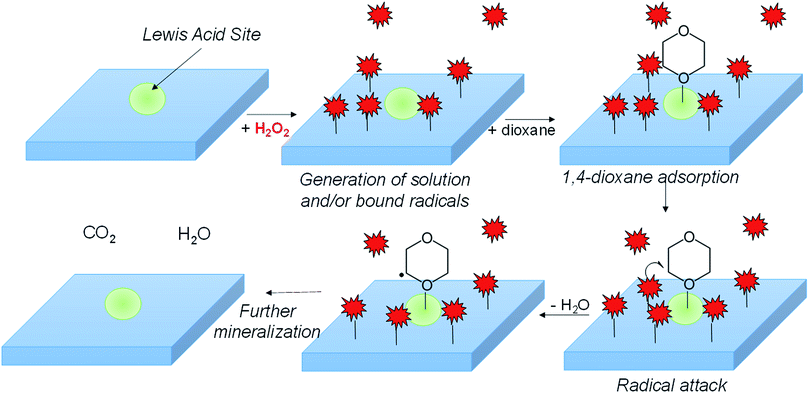
1,4-dioxane, commonly used as a solvent stabilizer and industrial solvent, is an environmental contaminant and probable carcinogen. In this study, we explored the concept of using metal oxides to activate H2O2 catalytically at neutral pH in the dark for 1,4-dioxane degradation. Based on batch kinetics measurements, materials that displayed the most suitable characteristics (high 1,4-dioxane degradation activity and high H2O2 consumption efficiency) were ZrO2, WOx/ZrO2, and CuO. In contrast, materials like TiO2, WO3, and aluminosilicate zeolite Y exhibited both low 1,4-dioxane degradation and H2O2 consumption activities. Other materials (e.g., Fe2O3 and CeO3) consumed H2O2 rapidly, however 1,4-dioxane degradation was negligible. The supported metal oxide WOx/ZrO2 was the most active for 1,4-dioxane degradation and had higher H2O2 consumption efficiency compared to ZrO2. In situ acetonitrile poisoning and FTIR spectroscopy results indicate different surface acid sites for 1,4-dioxane and H2O2 adsorption and reaction. Electron paramagnetic resonance measurements indicate that H2O2 forms hydroxyl radicals (˙OH) in the presence of CuO, and unusually, forms superoxide/peroxyl radicals (˙O2−) in the presence of WOx/ZrO2. The identified material properties suggest metal oxides/H2O2 as a potential advanced oxidation process in the treatment of 1,4-dioxane and other recalcitrant organic compounds.
131. H. Li, S. Guo, K. Shin, M.S. Wong, and G. Henkelman, "Design of a Pd–Au Nitrite Reduction Catalyst by Identifying and Optimizing Active Ensembles" ACS Catalysis 9, 9, 7957-7966 (2019) DOI:10.1021/acscatal.9b02182 (AIChE South Texas Section 2019 Best Fundamental Paper Award)
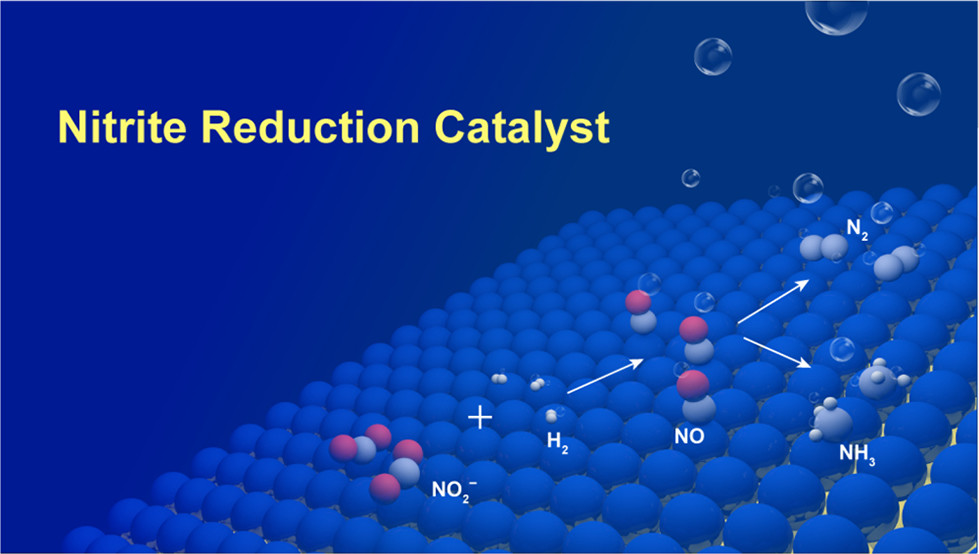
Nitrate (NO3–) is a ubiquitous contaminant in groundwater that causes serious public health issues around the world. Though various strategies are able to reduce NO3– to nitrite (NO2–), a rational catalyst design strategy for NO2– removal has not been found, in part because of the complicated reaction network of nitrate chemistry. In this study, we show, through catalytic modeling with density functional theory (DFT) calculations, that the performance of mono- and bimetallic surfaces for nitrite reduction can be rapidly screened using N, N2, and NH3 binding energies as reactivity descriptors. With a number of active surface atomic ensembles identified for nitrite reduction, we have designed a series of “metal-on-metal” bimetallics with optimized surface reactivity and a maximum number of active sites. Choosing Pd-on-Au nanoparticles (NPs) as candidate catalysts, both theory and experiment find that a thin monolayer of Pd-on-Au NPs (size: ~4 nm) leads to high nitrite reduction performance, outperforming pure Pd NPs and the other Pd surface compositions considered. Experiments show that this thin layer of Pd-on-Au has a relatively high selectivity for N2 formation, compared to pure Pd NPs. More importantly, our study shows that a simple model, based upon DFT-calculated thermodynamic energies, can facilitate catalysts design relevant to environmental issues.
130. Z. Zhao, K.N. Heck, P. Limpornpipat, H. Qian, J.T. Miller, M.S. Wong, "Hydrogen-generating behavior of Pd-decorated gold nanoparticles via formic acid decomposition" Catalysis Today 330, 24-31 (2019) DOI:10.1016/j.cattod.2018.06.044
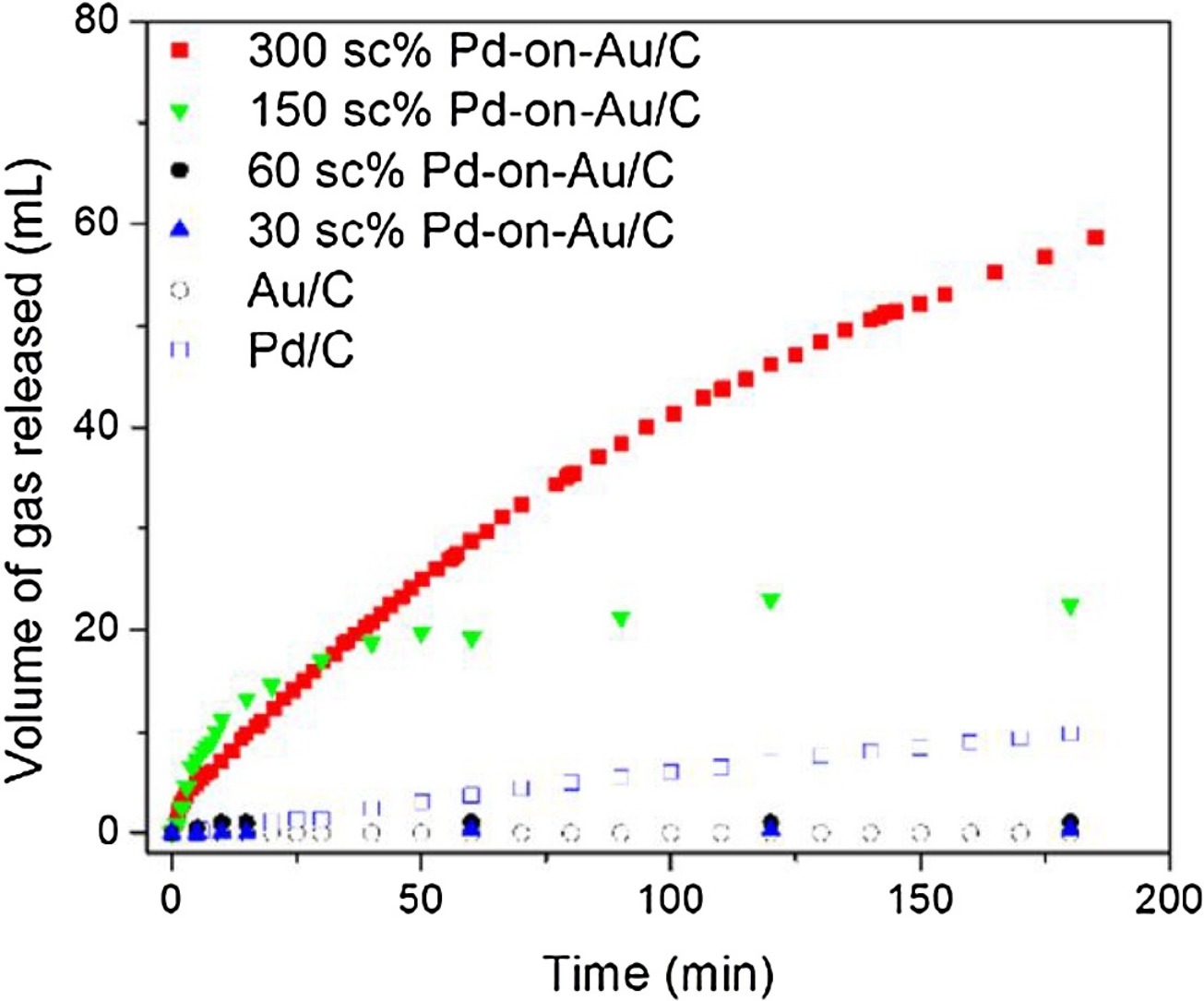
Formic acid is a promising hydrogen storage material where hydrogen is generated via metal-catalyzed decomposition. Bimetallic catalysts are active for this reaction, but the mechanism has not been fully proven. Palladium metal supported on gold nanoparticles (Pd-on-Au NPs) has structural properties that are advantageous for studying aqueous-phase catalytic reactions. In this work, a series of Pd-on-Au NPs of varying Pd loadings (calculated in terms of Pd surface coverage, sc%) were synthesized, immobilized onto carbon, and studied for formic acid decomposition at room temperature. Pd-on-Au NPs were catalytically active, with a reaction rate constant as high as 137 mL-H2/gPd/min (corresponding to an initial turnover frequency TOF of 123 h-1) at a Pd loading of 300 sc%. In contrast, Au NPs were inactive, and Pd NPs were slightly active (5 mL-H2/gPd/min and TOF of 38 h-1). The Pd metal of Pd-on-Au catalysts are partially oxidized, and is readily reduced without changing the metal-on-metal structure during reaction, according to in situ x-ray adsorption spectroscopy measurements. CO formation was inhibited at a Pd loading of 300 sc%, suggesting that three-dimensional Pd ensembles favored the desired dehydrogenation pathway while single-atom and small two-dimensional Pd ensembles are active for the undesired dehydration pathway.
129. Y.B. Yin, C.L. Conrad, K.N. Heck, F. Lejarza, and M.S. Wong, "Microencapsulated Photoluminescent Gold for ppb-level Chromium(VI) Sensing" ACS Applied Materials and Interfaces 11, 19, 17491-17500 (2019) DOI:10.1021/acsami.9b04699
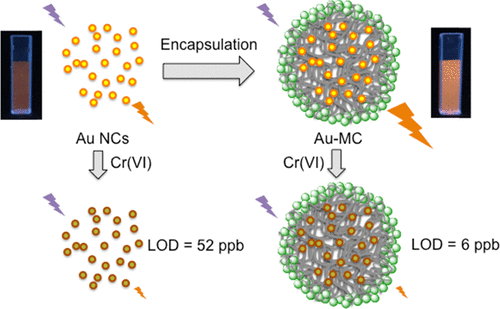
Luminescent gold nanoclusters (Au NCs) are a promising probe material for selective chemical sensing. However, low luminescent intensity and an incomplete understanding of the mechanistic origin of the luminescence limit their practical implementation. We induced glutathione-capped Au NCs to aggregate within silica-coated microcapsular structures using polymer-salt aggregate (PSA) self-assembly chemistry. The encapsulated NCs have a 5× luminescence enhancement compared to free Au NCs, and can detect Cr(VI) at concentrations as low as 6 ppb (= 0.12 µM CrO42-) through luminescence quenching, compared to free Au NCs which have a limit of detection (LOD) of 52 ppb (= 1 μM CrO42-). The LOD is 16× lower than the US EPA maximum contaminant level for total chromium (Cr(III) + Cr(VI), 100 ppb) in drinking water. No pH adjustment is needed using the encapsulated Au NCs, unlike the case for free Au NCs. The luminescent microcapsule material can sense Cr(VI) in simulated drinking water with a ~20-30 ppb LOD, serving as a possible basis for a practical Cr(VI) sensor.
128. T. Zhang, G.V. Lowry, N.L. Capiro, J. Chen, W. Chen, Y. Chen, D.D. Dionysiou, D.W. Elliott, S. Ghoshal, T. Hofmann, H. Hsu-Kim, J. Hughes, C. Jiang, G. Jiang, C. Jing, M. Kavanaugh, Q. Li, S. Liu, J. Ma, B. Pan, T. Phenrat, X. Qu, X. Quan, N. Saleh, P.J. Vikesland, Q. Wang, P. Westerhoff, M.S. Wong, T. Xia, B. Xing, B. Yan, L. Zhang, D. Zhou, and P.J.J. Alvarez, "In situ remediation of subsurface contamination: opportunities and challenges for nanotechnology and advanced materials" Enviornmental Science: Nano 6, 1283-1302 (2019) DOI:10.1039/C9EN00143C (Cover article) (ES Nano Best Paper Collection)
Complex subsurface contamination domains and limited efficacy of existing treatment approaches pose significant challenges to site remediation and underscore the need for technological innovation to develop cost-effective remedies. Here, we discuss opportunities for nanotechnology-enabled in situ remediation technologies to address soil and groundwater contamination. The discussion covers candidate nanomaterials, applications of nanomaterials to complement existing remediation approaches and address emerging contaminants, as well as the potential barriers for implementation and strategies and research needs to overcome these barriers. Promising nanomaterials in subsurface remediation include multi-functional nanocomposites for synergistic contaminant sequestration and degradation, selective adsorbents and catalysts, nano-tracers for subsurface contaminant delineation, and slow-release reagents enabled by stimuli-responsive nanomaterials. Limitations on mixing and transport of nanomaterials in the subsurface are severe constraints for in situ applications of these materials. Mixing enhancements are needed to overcome transport limitations in laminar flow environments. Reactive nanomaterials may be generated in situ to remediate contamination in low hydraulic conductivity zones. Overall, nano-enabled remediation technologies may improve remediation performance for a broad range of legacy and emerging contaminants. These technologies should continue to be developed and tested to discern theoretical hypotheses from feasible opportunities, and to establish realistic performance expectations for in situ remediation techniques using engineered nanomaterials alone or in combination with other technologies.
127. C.A. Clark, K.N. Heck, C.D. Powell, and M.S. Wong, "Highly Defective UiO-66 Materials for the Adsorptive Removal of PFOS" ACS Sustainable Chemistry & Engineering 7, 7, 6619-6628 (2019) DOI:10.1021/acssuschemeng.8b05572 (Virtual Special Issue)
Perflorooctane sulfonate (PFOS) is a persistent organic pollutant that is bioaccumulative and toxic. While its use in most countries has been restricted to certain industrial applications due to environmental and health concerns, chrome plating and semiconductor manufacturing facilities are industrial point sources of PFOS-containing wastewater. Current remediation technologies are ineffective at treating these highly concentrated industrial effluents. In this work, UiO-66 metal-organic frameworks (MOFs) of several defect concentrations were studied as sorbents for the removal of PFOS from concentrated aqueous solutions. PFOS sorption isotherms indicated that defective UiO-66, prepared with HCl as a modulator, had a maximum Langmuir sorption capacity of 1.24 mmol/g, which was ~2× greater than powdered activated carbon (PAC), but ~2× less than that of a commercial ion exchange resin. Defective UiO-66 adsorbed PFOS two orders of magnitude faster than the ion exchange resin. Large pore defects (~16 and ~20 Å) within the framework were critical to the increased adsorption capacity due to higher internal surface area and an increased number of coordinatively unsaturated Zr sites to bind the PFOS head groups. Of the common co-contaminants in chrome plating wastewaters, chloride ions have a negligible effect on PFOS sorption, while sulfate and hexavalent chromium anions compete for cationically charged adsorption sites. These materials were also effective adsorbents for the shorter-chain homologue, perfluorobutane sulfonate (PFBS). The enhanced PFOS and PFBS adsorptive properties of UiO-66 highlight the advantage of structurally defective MOFs as a water treatment approach towards environmental sustainability.
126. K.N. Heck, S. Garcia-Segura, P. Westerhoff, and M.S. Wong, "Catalytic Converts for Water Treatment" Accounts of Chemical Research 52, 4, 906-915 (2019) DOI:10.1021/acscatal.7b01371
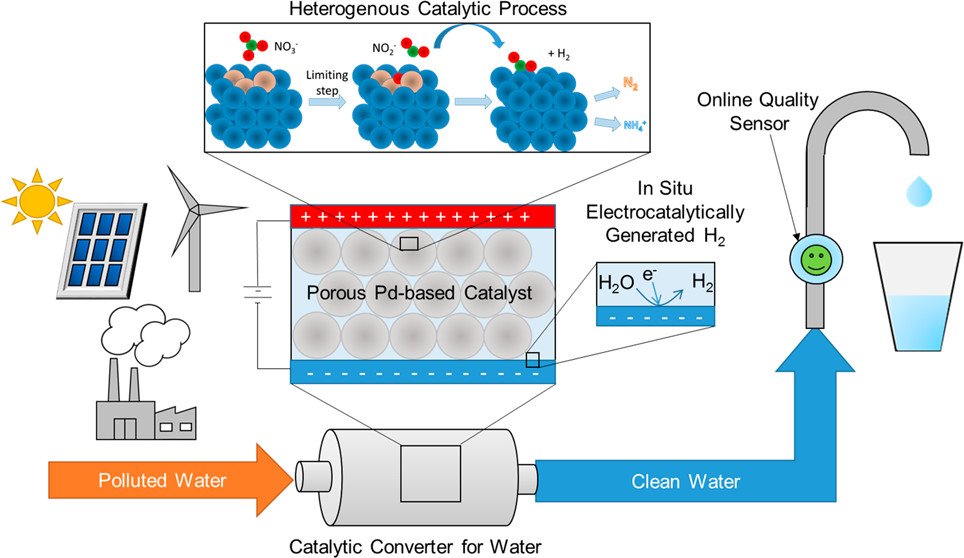
Fresh water demand is driven by human consumption, agricultural irrigation, and industrial usage and continues to increase along with the global population. Improved methods to inexpensively and sustainably clean water unfit for human consumption are desired, particularly at remote or rural locations. Heterogeneous catalysts
offer the opportunity to directly convert toxic molecules in water to nontoxic products. Heterogeneous catalytic reaction processes may bring to mind large-scale industrial production of chemicals, but they can also be used at the small scale, like catalytic converters used in cars to break down gaseous pollutants from fuel combustion. Catalytic
processes may be a competitive alternative to conventional water treatment technologies. They have much faster kinetics and are less operationally sensitive than current bioremediation-based methods. Unlike other conventional water treatment technologies (i.e., ion exchange, reverse osmosis, activated carbon filtration), they do not transfer
contaminants into separate, more concentrated waste streams.
In this Account, we review our efforts on the development of heterogeneous catalysts as advanced reduction technologies to treat toxic water contaminants such as chlorinated organics and nitrates. Fundamental understanding of the underlying chemistry of catalytic materials can inform the design of superior catalytic materials. We discuss the impact
of the catalytic structure (i.e., the arrangement of metal atoms on the catalyst surface) on the catalyst activity and selectivity for these aqueous reactions. To explore these aspects, we used model metal-on-metal nanoparticle catalysts along with state-of-the-art in situ spectroscopic techniques and density functional theory calculations to deduce
the catalyst surface structure and how it affects the reaction pathways and hence the activity and selectivity. We also discuss recent developments in photocatalysis and electrocatalysis for the treatment of nitrates, touching on fundamentals and surface reaction mechanisms.
Finally, we note that despite over 20 years of growing research into heterogeneous catalytic systems for water contaminants, only a few pilot-scale studies have been conducted, with no large-scale implementation to date. We conceive of modular, on- or off-grid catalytic units that treat drinking water at the household tap, at a community well, or
for larger-scale reuse of agricultural runoff. We discuss how these may be enhanced by combination with photocatalytic or electrocatalytic processes and how these reductive catalytic modules (catalytic converters for water) can be coupled with other modules for the removal of potential water contaminants.