Reviews
in Undergraduate Research - Issue 1
| CANCER
AND THE ROLE OF CELL CYCLE CHECKPOINTS
}
Will Renthal
University of Texas - Austin
Communicated By: Dr. Eva Lee
University of Texas Health Science Center at San Antonio, Institute
of Biotechnology |
SUMMARY
There exists an intricate network of proteins that continuously monitor
each phase of the cell cycle to ensure proper replication. This network
of proteins, termed checkpoints, first detects cellular abnormalities,
and then coordinates their repair before the cell divides. The malfunction
of these checkpoints often results in the proliferation of potentially
damaged cells, and thus a tremendous susceptibility to cancer. This
review will focus on the mechanisms by which checkpoints prevent the
proliferation of damaged cells through each phase of the cell cycle,
and how this understanding can provide novel targets for anticancer
therapy.
INTRODUCTION
The classic definition of cancer is "uncontrolled cell division."
In a large, multi-cellular organism, uncontrolled cell division will
soon result in large masses of rapidly growing cells (tumors), which
causes significant damage to surrounding tissues. When tumors spread,
they can damage vital organs and eventually cause death. In fact,
cancer is currently the second leading cause of death in the United
States, and thus cures for it would be of incalculable value. Current
treatments of cancer involve exposing the patient to relatively nonspecific
toxins, chemotherapy, in the hope that it will kill more cancer cells
than normal cells. This type of medicine is a modern equivalent of
18th century bleeding treatments for bacterial infections. However,
if clear biochemical differences between cancer cells and normal cell
are discovered, chemotherapy could be improved considerably. Much
as antibiotics only harm bacteria, novel anticancer drugs that only
harm cancer cells can be developed through research. Since cancer
is essentially the loss of cell division control, it seems prudent
to search in these regulatory mechanisms for distinguishing characteristics
of cancer cells. This review will present the general mechanisms which
drive the cell cycle and what is currently known about the regulatory
pathways that control it. It will then discuss how current anticancer
therapies are taking advantage of cell cycle research.
THE CELL CYCLE
The Nobel Prize in Medicine or Physiology was recently awarded to
three men, Leland Hartwell, Tim Hunt, and Paul Nurse, "for their
discoveries of key regulators of the cell cycle" (www.nobel.se).
Essentially every topic discussed in this review was in some way pioneered
by these three men. The details they helped uncover may seem at first
glance rather cumbersome, but it is these very details that will eventually
enable the development of targeted anticancer therapies.
Many of our cells can be triggered to divide upon the proper mitogenic
stimulus (Cross and Dexter, 1991). A growth factor for example, can
trigger a highly regulated unidirectional program that, when run successfully,
results in the proper replication and division of the cell. This program,
the cell cycle, must copy the parent cell's chromosomes and seal them
in a safe daughter cell with all of the essential components needed
for the daughter cell to function on its own. The human cell cycle
accomplishes this in approximately 24 hours through four major phases:
G1 (gap 1), S (DNA Synthesis), G2 (gap 2), and M (mitosis) (Lodish
et al., 2000). Each phase serves a specific function to ensure proper
cell division.
G1 - Gap 1
G1 takes about 9 hours to prepare the cell for DNA synthesis (S-phase)
(Lodish et al., 2000). Though preparing the cell for S-phase may
seem relatively uneventful, G1 is actually the most important regulatory
phase in the in the cell cycle. It is in this phase that the cell
decides whether to irreversibly continue the cell cycle through
mitosis, or to enter G0, a quiescent phase during which the cell
can function but not divide (Figure 1). This decision, called the
restriction point in mammalian cells, is made in late G1 based predominantly
on external growth signals (Lodish et al., 2000). Another key feature
of late G1, which contributes to the unidirectionality of the cell
cycle progression, is the priming of replication origins with MCM
(minichromosome maintenance) proteins. The binding of these proteins
to origins of replication is required for the initiation of DNA
synthesis, but they can only bind to DNA in late G1 (Young and Tye,
1997). Thus, DNA synthesis is only initiated once - right at the
G1/S transition.
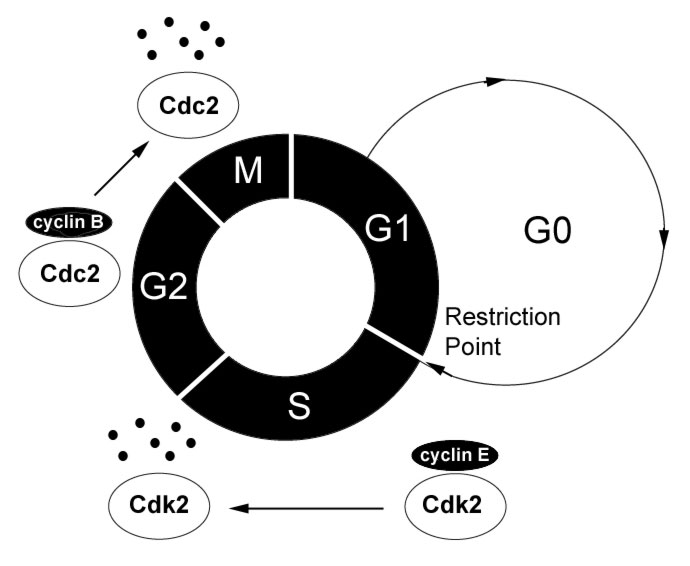
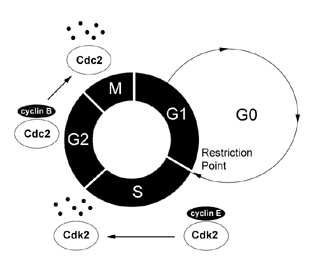 |
Figure
1: The Cell Cycle The
cell progresses through its division cycle, G1->S->G2->M,
in the presence of growth signals and active CDK/cyclin complexes
specific for each cell cycle stage. G0, a non-dividing but
functional phase, occurs in the absence of growth signals.
Finally, cyclin degradation is illustrated by the S-phase
cyclin/Cdk complex (cyclin E/Cdk2) and the G2/M cyclin/Cdk
complex (cyclin B/Cdc2), where each must be synthesized and
degraded systematically for proper cell cycle progression
into the next phase. [high
resolution pic]
|
S - DNA Synthesis
Once the cell has passed the restriction point, proteins synthesized
in late G1 initiate the DNA replication machinery of S-phase. This
delicate process of copying the parent cell's genome takes approximately
10 hours, and yields a single cell with two sets of each chromosome
(sister chromatids) (Lodish et al., 2000).
G2 - Gap 2
G2 lasts about 4.5 hours and serves as a buffer to ensure the completion
of DNA synthesis before the cell physically divides in mitosis (Lodish
et al., 2000). Now with two sets of each chromosome, cell growth
continues in order to double its size such that upon division two
fully functional cells will result. A large amount of information
is known about the G2/M transition, and it is discussed in detail
below. This phase serves to synthesize proteins required for nuclear
envelope breakdown, chromosome condensation, spindle formation,
and other processes required for entry into mitosis (Maller et al.,
1989). Many of these mitosis-promoting functions cannot be initiated
until DNA synthesis is completed, thus serving as a buffer phase
to prevent premature cell division.
M - Mitosis
Mitosis (nuclear division) is comprised of four substages during
which specific events occur to separate daughter chromosomes from
the parent's and enclose them in a new cell. Mitosis typically takes
about 30 minutes in human cells, which is rather fast considering
the complexity of this phase (Lodish et al., 2000). The first substage
to occur is Prophase, in which the proteins synthesized in G2 break
down the nuclear envelope of the parent cell, condense its chromosomes,
and initiate spindle formation. Prometaphase follows as a transition
period during which the sister chromatids shuffle until they align
in the middle of the cell, which is then termed Metaphase. The sister
chromatids then separate to opposite poles of the cell in Anaphase,
which is followed its physical division in Telophase.
Cyclins and Cyclin-Dependent
Kinases
The proper transition from each cell cycle phase to the next is
dependent on two classes of proteins called cyclins and cyclin-dependent
kinases. As the name suggests, cyclins are a class of proteins which
are periodically synthesized and degraded, and the coordination
of proper cyclin levels at the right time is essential for successful
cell cycle progression. Cyclin-dependent kinases (Cdk) are a class
of kinases whose catalytic activity is dependent on complexing with
an appropriate cyclin. Once this complex is formed, the Cdk kinase
activity is activated which results in the phosphorylation of many
downstream effectors (reviewed in Udvardy, 1996). This phosphorylation
serves to regulate the activity of the downstream effector, typically
by activating or inhibiting it. One of the best studied cyclin/Cdk
complexes involves cyclin B and Cdc2, also called MPF (Maturation
Promoting Factor), and was first discovered by Yoshio Masui and
Clement Market (Masui and Markert, 1971). It has been shown to serve
many crucial roles in cell cycle progression such as nuclear envelope
degradation and sister chromatid condensation in early mitosis (Maller
et al., 1989) (Figure 1). These MPF-dependent processes are essential
for the cell to efficiently divide the genetic information into
daughter cells, and thus the activity of MPF is tightly regulated.
If the cell allowed MPF to remain active through the latter stages
of mitosis when the nuclear envelopes are reforming, cell division
would be prevented altogether. The way the cell deals with this
problem is by ubiquitin-mediated proteolysis of cyclin B in late
mitosis (Murray et al., 1989). Thus, the actively regulated levels
of cyclin B mediate mitotic entry and exit. Without the synthesis
of cyclin B prior to the G2/M transition, the kinase activity of
Cdc2 remains inactive, and the cell can not enter mitosis. Without
the subsequent degradation of cyclin B, the kinase activity of Cdc2
remains active and prevents the exit from mitosis.
CELL CYCLE CONTROL:
CHECKPOINTS
A cell cycle checkpoint is a general term used to describe a cellular
process that stops or slows the cell cycle in conditions unfavorable
for cell division (Hartwell and Weinert, 1989). This review will focus
on the DNA damage cell cycle checkpoint. Since our cells undergo continuous
bombardment by DNA damaging agents such as UV light and by-products
of cellular metabolism, there exists an elaborate and evolutionarily
conserved cellular DNA damage response that coordinates cell cycle
progression with the repair of potentially mutagenic DNA damage (reviewed
in Zhou and Elledge, 2000). Many of the proteins involved in the mammalian
DNA damage response act as tumor suppressors and suggest that there
are newly evolved repair and/or checkpoint genes critical in the maintenance
of genome integrity, which highlights an additional importance of
checkpoint control in mammals. The DNA damage response, like most
cellular signaling pathways, involves first sensing a signal and then
transducing it to downstream effectors that elicit the appropriate
response. This review will present the recent studies, including some
previously unreviewed, which have provided tremendous insight into
many key components of this signal transduction pathway (outlined
in Table 1).
Table1:
Components of the DNA damage response
Cell cycle checkpoint proteins and their respective functions.
Putative, but not yet proven functions are followed by question
marks. |
| Name |
Function |
| PCNA |
Trimeric
DNA clamp, holds pols on DNA |
| RFC1-5 |
Pentameric
complex, loads PCNA onto DNA |
| CSC
- Checkpoint Sliding Clamp(Rad1, Rad9, Hus1) |
DNA
damage sensor? Structurally similar to PCNA |
| Rad17 |
DNA
damage sensor? Complexes with RFC2-5 to load CSC onto DNA |
| Rad26
(fission yeast)
ATRIP (humans) |
DNA
damage sensor? |
Rad3
(fission yeast)
ATR (humans) |
DNA
damage transducer |
| ATM |
DNA
damage transducer |
| p53 |
Transducer
for G1/S checkpoint and apoptosis |
| p21 |
Cdk4,6
Inhibitor, involved in G1/S checkpoint |
| Cdc25A |
Initiation
of DNA synthesis, S-phase checkpoint |
| Nbs1 |
S-phase
checkpoint transducer, DNA repair |
| Chk1 |
G2/M
checkpoint transducer |
| Cdc25C |
G2/M
checkpoint transducer |
| MPF
- Mitosis Promoting Factor(Cyclin B/Cdc2) |
Necessary
for G2/M transitionDNA damage effector, G2/M checkpoint |
| Cyclin
D/Cdk4,6 |
Necessary
for G1/S transitionDNA damage effector, G1/S checkpoint |
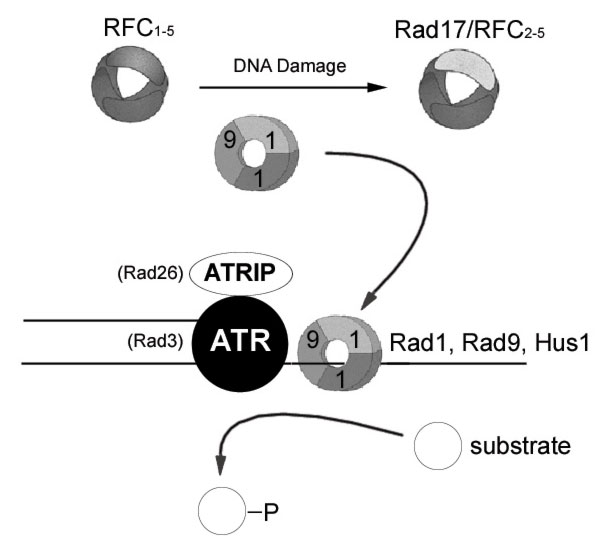
Sensors
It is still unclear precisely how the cell senses damaged DNA, but
a group of four fission yeast checkpoint proteins, Rad1, Rad9, Hus1,
and Rad17, have been implicated along with their respective homologues
in other organisms (reviewed in Lowndes and Murguia, 2000). It has
been proposed that Rad1, Rad9, and Hus1 form a trimeric checkpoint-sliding
clamp (CSC) similar in structure to the DNA polymerase clamp PCNA
(Proliferating Cell Nuclear Antigen) (Venclovas and Thelen , 2000).
By analogy to the loading of PCNA onto DNA by the RFC1-5 pentamer,
the CSC is loaded onto DNA by Rad17/RFC2-5, where Rad17 is a checkpoint
protein that replaces RFC1 in the pentamer (Venclovas and Thelen
, 2000). Once the CSC is loaded onto DNA, it could serve not only
to recruit DNA polymerase but also to signal the activation of downstream
DNA damage checkpoint proteins. In addition to the CSC and Rad17,
the checkpoint protein Rad26 has also been implicated as a DNA damage
sensor because it binds to and is phosphorylated by the Rad3 kinase
independently of all other checkpoint proteins (Edwards et al.,
1999). Thus, Rad26 has been proposed to be a sensor or at least
far upstream in the DNA damage response. A recent model, the substrate
recruitment model (Melo et al., 2001), provides some insight into
the behavior of the putative DNA damage sensors Rad26/Rad3 and CSC/Rad17
in budding yeast (Figure 2). The model proposes that each complex
is independently recruited to the same sites of DNA damage, and
work in tandem for proper DNA damage checkpoint activation (Kondo
et al., 2001; Melo et al., 2001). After the budding yeast CSC homologue
is loaded onto DNA near sites of damage, it recruits various substrates
of its Rad3 homologue to drive the signal transduction pathway of
the DNA damage response. Though this model seems quite convincing
in budding yeast, little is known about DNA damage sensing in mammalian
cells (Melo et al., 2001). Recently, it was shown that the activation
of both G1/S and G2 DNA damage checkpoints requires the phosphorylation
of hRad17 by ATR (a Rad3 related kinase in mammals) and possibly
ATM as well (Bao et al., 2001; Post et al., 2001). However, unphosphorylated
hRad17 can still load hRad9 of the CSC onto chromatin (Zou et al.,
2002), so the significance of this phosphorylation remains unclear.
Also, a recently cloned human protein, ATRIP, has some homology
to the putative DNA damage sensor Rad26 (Cortez et al., 2001). ATRIP
also seems to have many functional similarities to Rad26 including
its tight association with and its phosphorylation by ATR (Rad3
related kinase), and a role in the G2/M checkpoint. Thus, human
homologues of both of the putative DNA damage sensors in yeast seem
to play similar roles in the human DNA damage response.
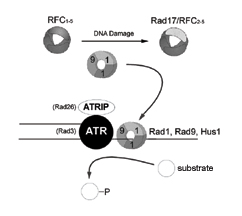 |
Figure
2: Substrate recruitment model for sensing DNA damage
Both ATRIP
(Rad26) and the checkpoint sliding complex (CSC) Rad1, Rad9,
Hus1, bind to the same sites of DNA damage. The CSC is loaded
onto chromatin after DNA damage in a Rad17/RFC2-5 catalyzed
reaction. Once chromatin bound, the CSC recruits substrates
of the ATR (Rad3) signal transducer kinase to propagate the
DNA damage response. [high
resolution pic]
|
Transducers
Once DNA damage is sensed, the cell must transduce this signal down
to its appropriate effector. In human cells, the activation of two
kinases is essential for the proper transduction of DNA damage:
ATM (Ataxia Telangiectasia Mutated) and ATR (ATM and Rad3 Related)
(reviewed in Elledge, 1996; Zhou and Elledge, 2000). Little is known
about precisely how ATM and ATR are activated, but a great deal
has been uncovered about their respective roles in coordinating
the DNA damage response. ATM was identified from a rare mutation
found in the disease ataxia telangiectasia and leads to chromosomal
instability and a high susceptibility to cancer (Savitsky et al.,
1995). There are no known pathologies with ATR mutations, and ATR
knockout mice die in early embryogenesis (Brown and Baltimore, 2000;
de Klein et al., 2001). Upon activation, these similar kinases phosphorylate
a number of target proteins, which transmit the DNA damage signal
downstream, eventually arresting the cell cycle, initiating DNA
repair, or, if necessary, causing cell death (apoptosis) (reviewed
in Zhou and Elledge SJ, 2000) (Figure 3).
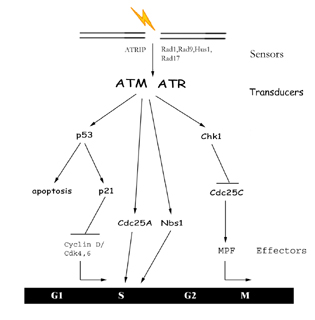 |
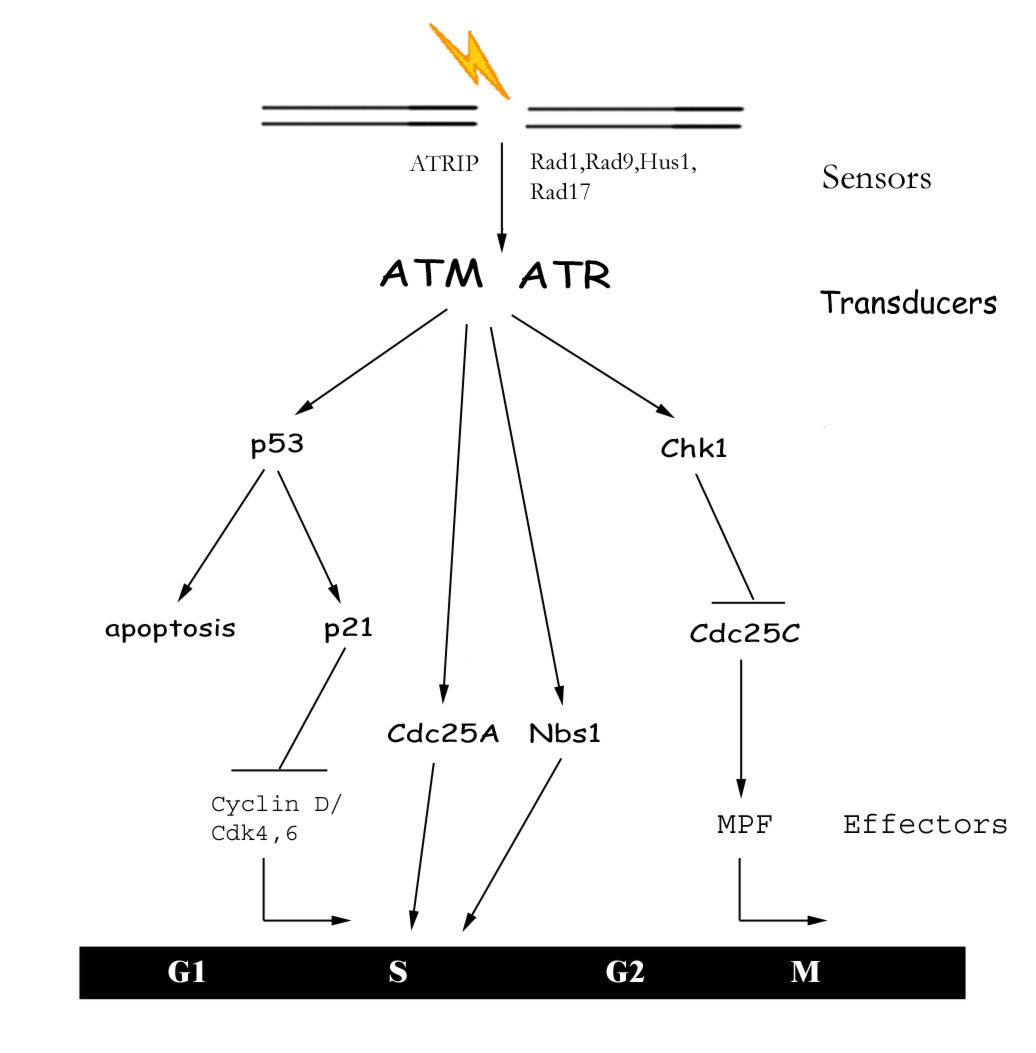
Figure
3: The DNA Damage Response DNA
damage is first detected by sensor proteins which in turn
activate transducers in the signal cascade. These transducers
then mediate the activation or inhibition of downstream effectors
which can arrest the cell cycle or cause apoptosis.
[high
resolution pic]
|
Depending on when the
DNA damage is sensed, ATM/ATR will activate a different axis of
proteins. For G1 damage, ATM/ATR will phosphorylate p53, which then
acts as a transcription factor for the synthesis of p21 (Cdk inhibitor)
(Canman, et al., 1998; Banin et al., 1998; Tibbetts et al., 1999).
Upon p21 expression, the G1/S transition is inhibited, preventing
the synthesis of damaged DNA (Li et al., 1994). If damaged DNA is
sensed during S-phase, the cell needs to slow DNA synthesis to provide
time for repair. An ATM dependent pathway exists for an intra S-phase
checkpoint in which ATM activation leads to the degradation Cdc25A
(Falck et al., 2001). Since Cdc25A drives the initiation of DNA
synthesis (Vigo et al., 1999), its degradation would allow synthesis
to slow during S-phase and provide the necessary time for repair.
This intra-S phase checkpoint is also controlled by the ATM phosphorylation
of Nbs1 (Lim et al., 2000; Zhao et al., 2000; Wu et al., 2000),
but the precise molecular mechanism remains elusive. Interestingly,
Nbs1 is a protein involved in DNA repair as well, so this functional
link between ATM and Nbs1 provides evidence of a high level of coordination
between cell cycle progression and DNA repair. DNA damage incurred
after S-phase results in the activation of the G2/M checkpoint to
prevent entry into mitosis with damaged chromosomes. ATM/ATR also
mediate this pathway by phosphorylating Chk1 in response to DNA
damage (Chen et al., 1999; Zhao et al., 2001). This phosphorylation
of Chk1 appears to enhance its kinase activity, which in turn phosphorylates
Cdc25C (Sanchez et al., 1997). Cdc25C is inactivated by this phosphorylation
and can no longer mediate entry into mitosis.
These signal transduction pathways (Figure 3) are just a few examples
of the complex interacting network of proteins actually involved
in processing DNA damage signals. The main idea embedded in these
vast yet important details is that proteins (ATM/ATR) are activated
upon DNA damage, and then trigger phase-specific cell cycle arrest
and DNA repair.
Effectors
The halting of the cell cycle is typically elicited by deactivating
the Cyclin/Cdk complex involved in a specific phase transition (G1/S
or G2/M). For example, p21 which is synthesized in response to DNA
damage in G1, directly inhibits Cdk4,6 and thus prevents the transcription
of proteins required for DNA synthesis. The final effector in the
G2/M checkpoint is the CyclinB/Cdc2 complex (MPF) described earlier
as being essential for the transition from G2 into mitosis. Upon
DNA damage, the Cdc25C phosphatase can no longer remove inhibitory
phosphates from Cdc2, and thus prevents the CyclinB/Cdc2 complex
from breaking down the nuclear envelope, condensing chromosomes,
and other events that occur in early mitosis.
CELL CYCLE CONTROLLERS
AS ANTICANCER DRUG TARGETS
Thus far this review has focused on the details underlying the control
of cell division, but it is important not to lose sight of the exciting
applications of this knowledge. For example, about half of all tumors
have a damaged copy of the tumor-suppressor protein p53. Now that
much of the detailed mechanism by which p53 inhibits tumor growth
is understood, drugs can be developed to take advantage of its action.
There are several biotechnology companies currently attempting to
develop p53 therapies by reconstituting functional p53 back into tumor
cells. This would restore the broken signal transduction pathway,
and thus prevent tumorigenesis. The DNA damage checkpoint consists
of many more pathways than those introduced in this review, and it
is difficult to say at this phase which pathways will be of greatest
utility in anticancer therapies. Thus, current cancer biology research
is directed at better characterizing known pathways and elucidating
novel ones involved in the DNA damage response.
ABOUT THE AUTHOR
Will Renthal is currently a third-year Biochemistry Honors student
at The University of Texas at Austin. There he conducts research as
an Arnold and Mabel Beckman Scholar on the mechanism by which a novel
antibiotic kills bacteria and as an NSF Fellow on MAP Kinase signal
transduction. For the previous two summers, he has researched cell
cycle checkpoints with Dr. Eva Lee at the University of Texas Health
Science Center at San Antonio, Institute of Biotechnology. His research
focused specifically on characterizing the functions of a protein
which is commonly mutated in Nijmegen Breakage Syndrome (NBS) patients.
This is a disease in which patients have an extremely high susceptibility
to cancer and chromosomal instability. In his two summers of research,
he has helped to clarify some of the subtle points about the NBS gene
product, which is involved in both cell cycle checkpoints and DNA
repair.
After his undergraduate education he plans to attend either an MD/PHD
program or graduate school where he will further study the mechanisms
underlying cell growth and development. Following this graduate training
and a brief postdoctoral position, he aspires to become a professor
at a medical center. There he hopes to conduct high quality basic
research with a focus on drug discovery while teaching the next generation
of scientists.
ACKNOWLEDGEMENTS
I would like to thank Dr. Eva Lee for providing me with two incredible
summers of exciting research; Dr. Song Zhao for serving as my mentor
and friend; Sean Post for stimulating discussions; and the rest of
the lab for their support. I also thank Drs. Ann and Robert Renthal
for their critical reading of this manuscript.
FURTHER READING
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, Keith., Walter,
P. Molecular Biology of the Cell, 4th edn. New York: Taylor
& Francis Group, 2002.
Dasika GK, Lin
SC, Zhao S, Sung P, Tomkinson A, Lee EY. (1999) DNA damage-induced
cell cycle checkpoints and DNA strand break repair in development
and tumorigenesis. Oncogene 18, 7883-7899.
Zhou BB, Elledge
SJ. (2000) DNA damage response: putting checkpoints in perspective.
Nature. 408, 433-439.
Gutkind, JS. Signaling
Networks and Cell Cycle Control: The Molecular Basis of Cancer and
Other Diseases. New York: Humana Press Inc., 2000.
REFERENCES
Alberts, B., Bray, D., Lewis,
J., Raff, M., Roberts, K., and Watson, J. Molecular Biology of
the Cell, 3rd edn. New York: Taylor & Francis Group, 1994.
Banin, S. et al. Enhanced
phosphorylation of p53 by ATM in response to DNA damage. (1998) Science
281, 1674-1677
Bao S, Tibbetts RS, Brumbaugh
KM, Fang Y, Richardson DA, Ali A, Chen SM, Abraham RT, Wang XF. (2001)
ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic
stress responses. Nature 411, 969-974.
Brown, EJ and Baltimore,
D. ATR disruption leads to chromosomal fragmentation and early embryonic
lethality. (2000) Genes Dev. 14, 397-402.
Canman, C. E. et al. Activation
of the ATMkinase by ionizing radiation and phosphorylation of p53.
(1998) Science 281, 1677-1679.
Chen P, Gatei M, O'Connell
MJ, Khanna KK, Bugg SJ, Hogg A, Scott SP, Hobson K, Lavin MF. (1999)
Chk1 complements the G2/M checkpoint defect and radiosensitivity of
ataxia-telangiectasia cells. Oncogene 18, 249-256.
Cortez D, Guntuku S, Qin
J, Elledge SJ. (2001) ATR and ATRIP: Partners in Checkpoint Signaling.
Science 294, 1713-1716.
Cross M, Dexter TM. (1991)
Growth factors in development, transformation, and tumorigenesis.
Cell 64, 271-280.
de Klein, A. et al. Targeted
disruption of the cell-cycle checkpoint gene ATR leads to early embryonic
lethality in mice. (2000) Curr. Biol. 10, 479-482.
Edwards RJ, Bentley NJ,
Carr AM. (1999) A Rad3-Rad26 complex responds to DNA damage independently
of other checkpoint proteins. Nat Cell Biol. 7, 393-398.
Elledge SJ. (1996) Cell
cycle checkpoints: preventing an identity crisis. Science 274, 1664-1672.
Falck J, Mailand N, Syljuasen
RG, Bartek J, Lukas J. (2001) The ATM-Chk2-Cdc25A checkpoint pathway
guards against radioresistant DNA synthesis. Nature 410, 842-847.
Hartwell LH, Weinert TA.
(1989) Checkpoints: controls that ensure the order of cell cycle events.
Science 246, 629-634.
Kondo T, Wakayama T, Naiki
T, Matsumoto K, Sugimoto K. (2001) Recruitment of Mec1 and Ddc1 checkpoint
proteins to double-strand breaks through distinct
mechanisms. Science 294, 867-870.
Li Y, Jenkins CW, Nichols MA, Xiong Y. (1994) Cell cycle expression
and p53 regulation of the cyclin-dependent kinase inhibitor p21. Oncogene
9, 2261-2268.
Lim DS, Kim ST, Xu B, Maser
RS, Lin J, Petrini JH, Kastan MB. (2000) ATM phosphorylates p95/nbs1
in an S-phase checkpoint pathway. Nature 404, 613-617.
Lodish, H., Ber, A., Zipursky,
LS., Matsudaira, P., Baltimore, D., Darnell, J. Molecular Cell
Biology. New York: W. H. Freeman and Company, 2000.
Lowndes, N. and Murguia,
J. (2000) Sensing and responding to DNA damage. Curr. Opin. Genet.
Dev. 10, 17-25.
Maller Jl, Gautier J, Langan
TA, Lohka MJ, Shenoy S, Shalloway D, Nurse P. (1989) Maturation-promoting
factor and the regulation of the cell cycle. J. Cell Sci. Suppl. 12,
53-63.
Masui, Y. and Markert,
CL. (1971) Cytoplasmic Control of Nuclear Behavior during Meiotic
Maturation of Frog Oocytes. J. Exp. Zoology, 177, 129-146.
Melo JA, Cohen J, Toczyski
DP. (2001) Two checkpoint complexes are independently recruited to
sites of DNA damage in vivo. Genes Dev. 21, 2809-2821.
Murray AW , Solomon MJ
, Kirschner MW. (1989) The role of cyclin synthesis and degradation
in the control of maturation promoting factor activity. Nature 339,
280-286.
Post S, Weng YC, Cimprich
K, Chen LB, Xu Y, Lee EY. (2001) Phosphorylation of serines 635 and
645 of human Rad17 is cell cycle regulated and is required for G(1)/S
checkpoint activation in response to DNA damage. Proc. Natl. Acad.
Sci. USA 6, 13102-13107.
Sanchez Y, Wong C, Thoma
RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. (1997) Conservation
of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to
Cdk regulation through Cdc25. Science 277, 1450-1451.
Savitsky, K., Bar-Shira,
A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle, D.A., Smith,
S., Uziel, T., Sfez, S., et al. (1995) A single ataxia telangiectasia
gene with a product similar to PI-3 kinase. Science 268 1749-1753.
Tibbetts, R. S. et al.
A role for ATR in the DNA damage-induced phosphorylation of p53. (1999)
Genes Dev. 13, 152-157.
Udvardy A. (1996) The role
of controlled proteolysis in cell-cycle regulation. Eur. J. Biochem.
240, 307-313.
Venclovas, C. and Thelen,
M. (2000) Structure-based predictions of Rad1, Rad9, Hus1, and Rad17
participation in sliding clamp and clamp-loading complexes. Nucleic
Acids Res. 28, 2481-2193.
Vigo E, Muller H, Prosperini
E, Hateboer G, Cartwright P, Moroni MC, Helin K. (1999) CDC25A phosphatase
is a target of E2F and is required for efficient E2F-induced S phase.
Mol. Cell. Biol. 19, 6379-6395.
Wu X, Ranganathan V, Weisman
DS, Heine WF, Ciccone DN, O'Neill TB, Crick KE, Pierce KA, Lane WS,
Rathbun G, Livingston DM, Weaver DT. (2000) ATM phosphorylation of
Nijmegen breakage syndrome protein is required in a DNA damage response.
Nature 404, 477-482.
www.nobel.se
Young MR, Tye BK. (1997)
Mcm2 and Mcm3 are constitutive nuclear proteins that exhibit distinct
isoforms and bind chromatin during specific cell cycle stages of Saccharomyces
cerevisiae. Mol. Biol. Cell. 8, 1587-1601.
Zhao H, Piwnica-Worms H.
(2001) ATR-mediated checkpoint pathways regulate phosphorylation and
activation of human Chk1. Mol. Cell. Biol. 21, 4129-4139.
Zhao S, Weng YC, Yuan SS,
Lin YT, Hsu HC, Lin SC, Gerbino E, Song MH, Zdzienicka MZ, Gatti RA,
Shay JW, Ziv Y, Shiloh Y, Lee EY. (2000) Functional link between ataxia-telangiectasia
and Nijmegen breakage syndrome gene products. Nature 405, 473-477.
Zhou BB, Elledge SJ. (2000)
DNA damage response: putting checkpoints in perspective.
Nature. 408, 433-439.
Zou L, Cortez D, Elledge
SJ. (2002) Regulation of ATR substrate selection by Rad17-dependent
loading of Rad9 complexes onto chromatin. Genes and Development 16,
198-208.
|

{kind=link}
{kind=link}
{kind=link}