|
Reviews
in Undergraduate Research - Issue 1
| THE
ROLE OF MECHANICAL STRESS IN SKELETAL MYOCYTES: MAPK SIGNAL TRANSDUCTION
PATHWAYS
}
David A. Barron: Rice University
Dr. Ashok Kumar: Baylor College of Medicine
Dr. Aladin M. Boriek: Baylor Cellege of Medicine |
SUMMARY
The idea of signal transduction provides the necessary link between
a stimulus from the external environment of a cell, and the intricacies
that occur among its intracellular components. The proteins embedded
in the plasma membrane of a cell assist in this transduction in that
they act as molecular antennae, capturing the initiating external stimulus
and transmitting its effects to the cell cytoplasm. Albeit many molecules
can freely pass through the cell membrane, those that are effectively
excluded by this semipermeable barrier would have no way of communicating
with the cell interior without the process of signal transduction. Consequently,
many pivotal biochemical pathways would be completely hindered and the
life of the organism affected would cease. The mitogen-activated protein
kinase (MAPK) pathway is one of the most significant signaling systems
used by an organism to elicit a variety of responses at the cellular
level. This unique pathway is thought to be directly responsible for
regulating cell proliferation, differentiation, and survival, all of
which are vital for the existence of life. Even though the basic regulatory
steps have been delineated, many fascinating features of this pathway
are only beginning to emerge. The conventional study of signal amplification,
which has historically encompassed the analysis of ligand interaction
with cell membrane receptors, can be slightly modified to incorporate
mechanical stress as the initiating component in the signaling pathway.
Studying signal transduction in light of mechanical stretching may help
enhance the understanding of pathway-initiating mechanisms in a way
that can be applicable to mechanical manipulation of the extracellular
matrix and plasma membrane domains. Such manipulation is seen in vivo,
with normal physiological muscle contraction, as well as in vitro, with
concentric and eccentric muscle stretching maneuvers. Furthermore, tracing
the stimulus of cell stretching all the way to gene transcription demonstrates
that many proteins from different families are involved in facilitating
the activation of MAPKs. Theories on integrin signaling show how cell
stretching induces the activation of protein kinase C (PKC), leading
to the release of Ca2+ ions which are then sequestered by integrins.
These active integrins may then activate focal adhesion kinases (FAK),
which constitutes a major initiating pathway to activating MAPKs. Alternatively,
integrins may immediately be activated by their associations with the
extracellular matrix, which then activate FAK in a similar fashion.
The dominance of ionic over mechanical activation is still unknown,
and a combination of these mechanisms may contribute to the initiation
of MAPKs. Observation of cell spreading and growth, two characteristics
of activating MAPK pathways, can prove to verify the effects of mechanotransduction
in muscle stretching. While the hypothetical models presented here provide
a logical synthesis of concepts in signal transduction, the complexity
surrounding the field leads to the idea that an intricate interplay
of chemical processes within the world of the cell exists. The mediators
of these processes include, but are certainly not limited to, integrin
activation via calcium binding or dimerization of its subunits.
INTRODUCTION
A cell's decision to grow, proliferate, or terminate itself is the end
product of long and complex deliberations. For instance, a quiescent
(nongrowing) cell must receive and process a number of growth-stimulatory
signals, notably those conveyed by growth factors, and assess whether
its strength and number warrant entrance into an active proliferative
phase. Decision-making such as this demands a complex signal-processing
apparatus inside the cell. A helpful metaphor is an electronic circuit
board constructed as a network of components that operate like resistors,
transistors, and capacitors (Weinberg, 1998). Each of these components
is a logical device that receives signals from other components, processes
and interprets these signals, and then passes them on to other circuit
elements. In the living cell, circuit components like these are proteins
endowed with complex signal-processing capabilities. These proteins
are capable of 'signal transduction' in that they receive signals, filter
and amplify them, and then pass them on to other components. In this
way, signal transduction allows the cell's external environment to govern
intracellular machinery in an orderly fashion. Although there are many
substances that can physically traverse the plasma membrane barrier,
namely, hydrophobic substances and other relatively small molecules,
there are still quite a few instrumental molecules that are effectively
excluded by this barrier. Such substances would thus have no effect
on cytoplasmic elements without the mechanism that is known as signal
transduction. In this process, proteins function like molecular bucket
brigades (Weinberg, 1998). A protein at the top of this brigade relays
a signal to the next protein down the line, which in turn responds by
transmitting the signal yet another step down. Such chains of command
are commonly referred to as signal cascades. In actuality, the initiating
signal is amplified virtually exponentially, in that each component
that is activated in turn can activate several others to the extent
that the effects are seen on a global scale. Of the known contributors
to signaling cascades, mitogen activated protein kinases (MAPKs) are
among the most versatile known. MAPKs are evolutionary conserved enzymatic
complexes connecting cell membrane epitopes, or regions capable of provoking
a cellular response, to regulatory intracellular endpoints. They respond
to chemical and, as will be shown, mechanical stresses, in an effort
to control cell survival and adaptation. MAPK activity is regulated
through three-tiered cascades composed of a MAPK, a MAPK kinase (MEK),
and an MEK kinase (MEKK) (English et al, 1999). These modules may be
activated by small guanosine triphoshate (GTP) binding proteins, namely
via G-protein linked receptors (Gutkind, 2000). In theory, all MAPK
pathways activated in the forward direction, through substrate-level
phosphorylation, can be inactivated by MAPK phosphatases.
Four parallel cascades of MAPKs have recently been described in mammalian
cells, including extracellular signal-regulated kinase (ERK), stress-activated
protein kinase p38, and cJun NH2-terminal kinase (figure 1). An instrumental
factor involved in the ERK cascade is Raf, which is a serine/threonine
kinase that phosphorylates downstream targets in signaling pathways.
MAPKs are activated via dual phosphorylation on threonine and tyrosine
residues by MAPKs. Factors that have been shown to trigger MAPK activation
include hormones, growth factors, reactive oxygen species, lowered pH,
and mechanical stress (Widmann et al, 1999). Following activation, MAPKs
can either phosphorylate different cytoplasmic targets or translocate
to the nucleus and directly or indirectly affect transcription (Wretman
et al, 2001).
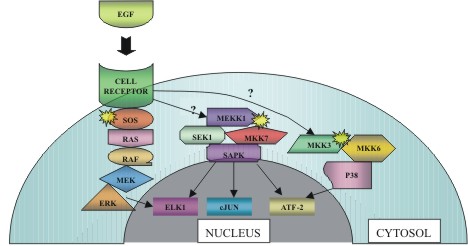 |
Figure
1: Hierarchy of MAPK signals.
Signals from the cell surface are transduced through the cytoplasm
by a cascade of protein kinases. In this case, epidermal growth
factor (EGF) acts as a signaling promoter by binding to its transmembrane
receptor domain with concomitant activation of Sos (the yellow sunburst
symbol representing activation will be used throughout this article).
The relationship between ligand binding and activation of both the
SAPK and p38 pathways is still unclear. (Figure adapted from L.A.
Tibbles, et. al.) |
The general pathway for stress activated protein kinases (SAPKs), which
originates from members of the MAPK family, involves Raf, MEK, and ERK.
The three main mediators of this pathway are ERK, SAPK, and p38, which
each eventually have an effect on transcription factors in the nucleus
(figure 1). As can be seen in figure 1, the ERK pathway is a hierarchical
cascade originating at the cell membrane with receptors for either mitogens,
which are substances that cause cells to undergo cell division, or growth
factors. These receptors recruit, via adaptor proteins and exchange
factors, the small guanosine triphosphatase (GTPase) known as Ras (Tibbles
et al, 1999). Ras then subsequently activates Raf, a serine threonine
kinase, which activates MEK (MAPK/ERK kinase). MEK, in turn, phosphorylates
and activates ERK1 and ERK2 which translocate to the nucleus perhaps
via a nuclear localization sequence (Alberts et al, 1994). These two
proteins transactivate transcription factors, changing gene expression
to promote cellular expansion, differentiation, or mitotic division.
It has been shown that signal transduction pathways are activated via
stress and inflammatory mechanisms in mammalian somatic cells (Kyriakis
et al, 2001). Environmental stresses such as physical exertion or mechanical
manipulation of the muscle tissue constitute external stimuli that may
employ MAPKs capable of initiating SAPKs (Tibbles et al, 2000). Both
of these categories of stress will be discussed in turn, including their
effects on the extracellular matrix and cytoskeletal agents.
As the regulation of mitogen activated cell signaling has been discussed
in detail elsewhere (English et al, 1999), this article focuses on contemporary
developments in understanding MAPK function in mammalian systems, coupled
with stress-related stimuli from the external environment. The analysis
of targeted mutations in mice and development of specific inhibitors
have contributed to a greater understanding of the definitive role of
MAPKs in mammals. It is becoming increasingly evident that MAPKs regulate
almost all cellular processes, from transcription of genetic information
to programmed cell death.
KEY COMPONENTS IN
MAPK ACTIVATION
The stress activated
protein kinases (SAPKs)
SAPKs reversibly bind and phosphorylate the transcription factor cJun
(figure1). cJun is one portion of the activator protein 1 (AP-1) transcription
factor complex; the remaining constituent parts include members of
the cFos and cJun superfamilies. Transactivation of cJun (figure 2)
by the SAPKs leads to increased expression of the genes that have
AP-1 regions in their promoters (Tibbles et al, 1999). Among the primary
targets of the AP-1 is the cJun gene itself, so transactivation of
cJun initiates a positive feedback loop (Tibbles et al, 1999) which
is presumably terminated upon transcription of the cJun gene. As can
be seen in Figure 1, SAPK effectively acts as a universal pivot point,
with targets to both a ternary complex transcription factor (ELK-1)
and activating transcription factor 2 (ATF-2). The ternary complex
factor ELK-1, once activated by SAPK, leads to positive regulation
of the cFos promoter resulting in increased expression of the cFos
protein with concomitant increases in AP-1 levels (Tibbles et al,
1999) (figure 2). Targeting of ATF-2, which can form heterodimers
with cJun, is another suitable route to initiate increases in AP-1
expression. Given the myriad of possibilities for activating AP-1,
it is quite apparent that the SAPK is a model transduction junction
for amplifying a given extracellular signal. The SAPKs are encoded
by at least three genes, and as with all MAPKs, each SAPK isoform
contains a characteristic Thr-X-Tyr phosphoacceptor loop domain, where
X indicates any amino acid structurally suitable for a loop domain
(Kyriakis et al, 2001).
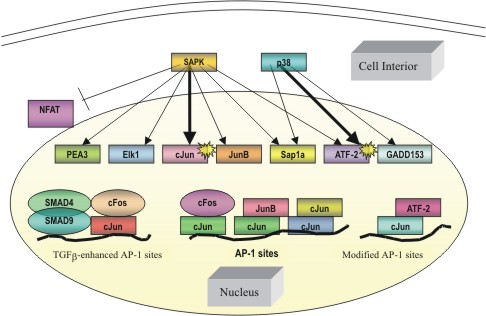 |
Figure
2: Transcription factor targets of the stress-response kinases.
SAPK activation via an extracellular signal leads to the phosphorylation
of specific downsteam transcription factors, leading to an effect
at the level of the gene. Various forms of these transcription
factors ultimately promote the transcription of genes with binding
sites for the AP-1 complex. Arrows in bold correspond to major
activation pathways outlined in Figure 1. |
p38 protein kinases
p38 kinases respond to virtually the same agonists that activate the
structurally similar SAPKs (i.e. members of the MAPK family), but
under certain circumstances they are differentially regulated (Mendelson
et al, 1996). As seen in figure 2, they activate via phosphorylating
the transcription factor ATF-2 as well as growth arrest and DNA damage
transcription factors (Tibbles et al, 1999). As will be discussed
later, p38 activation can be mediated by protein kinase C (PKC) by
an experimentally unidentified mechanism (Ryder et al, 2000). Muscle
contraction has been implicated in activation of PKC in response to
electrical stimulation, however (Richter et al, 1988). As a checkpoint
prior to nuclear translocation, p38 appears to be instrumental in
regulating a variety of cellular processes, ranging from maintenance
of genetic information to preservation of the cell line.
ERK- a third class
of Stress-Activated MAP Kinases
The extracellular regulated protein kinases (ERK), with a phosphoacceptor
sequence of Thr-Glu-Tyr, contain an NH2-terminal kinase domain followed
by an extensive COOH-terminal tail of unknown function that has several
proline-rich motifs indicative of binding sites with SH3 domains (Zhou
et al, 1995). These SH3 adaptor proteins are instrumental in linking
the initial activation of a kinase to the downstream components of
any signal transduction pathway. Although the stimuli that recruit
ERK kinases have not been well identified, environmental stresses
such as osmotic shock and oxidant stress have been shown to substantially
activate ERK and similar substrates (Abe et al, 1996). EGF activation
of ERK has been subsequently documented in studies done on cultured
cells (Chao et al, 1999) suggesting that a MAPK may be involved (figure
1).
EFFECTS OF MYOCYTE
STRETCHING ON MAPKS
As previously mentioned, there are a variety of environmental stimuli
that activate MAPK pathways. Of these stimuli, manipulation of the cell
plasma membrane and associated extracellular matrix appears to be especially
effective in propagating signal transduction pathways. Voluntary stretching
of muscle tissue, exercise induced muscle contraction, and inflammation
of muscle cell (myocyte) tissue are all examples of such manipulation
of the cell exterior. While many hypotheses and models for mechanotransduction
exist, the role of tensile and shear forces on activating MAPKs will
be the focus here.
Concentric and Eccentric
contractions
Studies on isolated rat skeletal muscle have shown an increase in
phosphorylation of both ERK and p38 MAPKs via mechanical alterations,
whereas an increase only in ERK activity was caused by contraction-related
metabolic/ionic changes (Wretman et al, 2001). The latter effectors
for ERK activation stem from acidotic cell conditions, in which the
buildup of lactic acid from cellular respiration induces, either directly
or indirectly, the activation of MAPKs. This idea will have some interesting
implications in exercise-induced stimulation of MAPKs, as will be
discussed in the following section.
Concentric muscle contractions (i.e. contractions induced in muscle
fibers along a common axis situated at the geometric center) have
been found to have divergent effects on MAPKs in that they induce
a marked elevation in ERK phosphorylation, whereas p38 is not significantly
affected (Wretman et al, 2001). In addition, the increase in phosphorylation
of ERK, but not p38, can be induced by metabolic changes, such as
acidification, that occur during repeated contractions and also by
mild mechanical perturbations (Wretman et al, 2001). Eccentric contractions
(i.e. contractions induced in muscle fibers along an axis other than
that situated at the geometric center), on the other hand, seem to
markedly facilitate the phosphorylation of both ERK and p38 MAPKs
(Wretman et al, 2001). Thus there seems to be an alternative mechanism
at work in concentric contractions that selectively stimulates the
initiation of ERK and not p38. It may be hypothesized that the concentric
contractions do not generate sufficient force to exert an effect on
the p38 pathway, which may give rise to the idea that the upstream
components of the p38 activation pathway are more internal to the
cell surface, in relation to the ERK elements. However, there still
remains much to be discovered about the nature of these concentric/eccentric
contractions and their relationship to the activation of these MAPK
components.
The fact that p38 MAPK phosphorylation is not affected greatly by
concentric contractions implies that it is little affected by metabolic
alterations, and previous studies have shown no effect on phosphorylation
by acidotic conditions (Wretman et al, 2001). Furthermore, since Wretman
has shown that p38 phosphorylation is not induced by mild mechanical
stress, by exclusion the higher mechanical stress imposed upon muscle
in isometric contractions is required to induce an increase in p38
MAPK phosphorylation. This was verified in Wretman's studies by the
finding that eccentric contractions markedly induced phosphorylation
of p38 MAPK, an effect that also tended to occur with severe stretching
maneuvers. The importance of mechanical stress in enhancing MAPK phosphorylation
is now becoming increasingly evident. As far as the mechanism underlying
MAPK phosphorylation with eccentric contractions is concerned, it
appears that a number of internal cytoskeletal elements are involved.
In eccentric contractions, the contractile units (cross bridges) and
the elastic elements in series (z-lines and tendons) are involved
in force generation and transmission, whereas in severe stretch, stiffness
of elastic components in parallel (sarcolemma, endomysium, perimysium,
and epimysium) and in series generates force (Wretman et al, 2001).
Exercise stimulation
It has been shown that exercise- and contraction-induced ERK signaling
involves the same Ras/Raf/MEK pathway in the activation of ELK1 (Aronson
et al, 1997) (figure 1). PKC activation is also known to lead to Ras
activation and thus stimulate MAPK activity (van Biesen et al, 1996).
Additionally, PKC can mediate p38 MAPK activation by an unidentified
mechanism. One possible mechanism may involve a signal cascade, in
which PKC phosphorylates a MAPK at its tyrosine and threonine phosphoacceptor
domains (figure 3). The activation of PKC occurs via a G-protein,
which is itself activated by the binding of an agonist ligand to its
specific cell receptor (figure 3). The mediator, phospholipase C,
cleaves phosphatidylinositol bisphosphate (PIP2) into diacylglycerol
(DAG) and inositol triphosphate (IP3), the former of which directly
activates PKC (Voet et al, 1995).
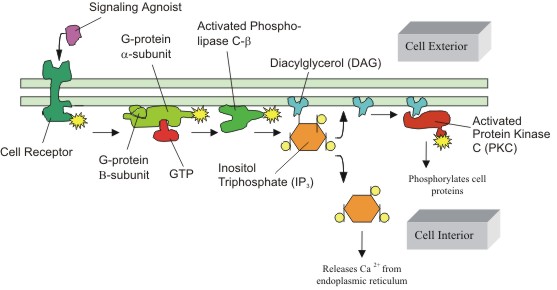 |
Figure
3: Activation of Protein Kinase C (PKC) via G-proteins.
The activation of PKC is preceded by a number of steps, originating
from the binding of an extracellular ligand that activates a G-protein
on the cytosolic side of the plasma membrane. The G-protein, using
GTP as an energy source, then activates PKC via the phosphatidylinositol
bisphosphate (PIP2) intermediate, which is shown as the DAG/IP3
complex. (Figure adapted from Alberts, B. et al) |
As previously mentioned,
the activation of PKC has been suggested to be caused by muscle contraction
in response to electrical stimulation (Cleland et al, 1989). Coupling
this idea with myocyte stretching might provide a relationship between
the presence of an external force and the binding of the activating
ligand. If one would imagine this ligand in the vicinity of the extracellular
matrix, the direction and magnitude of force in cell stretching would
become a major determining factor in the orientation and proper binding
of the agonist to its transmembrane receptor. This would support the
selective activation of p38 MAPKs only in eccentric contractions.
Moreover, the selective responsiveness of certain upstream elements
of MAPKs (i.e. PKC) to electrical stimulation may be the result of
ionic changes within the cell in vivo.
Energy considerations within a cell and acidification of the cytosolic
environment can also shed light on the regulation of MAPKs. When energy
turnover in contracting muscle is high, the intracellular milieu becomes
acidic due to the buildup of lactic acid discussed earlier (Aronson
et al, 2000). A high energy turnover is indicative of a high rate
of cellular respiration, which in turn is the result of active muscle
contraction during physical exertion. In vitro analysis of muscle
stretching indicates that acidosis, of the magnitude similar to acidosis
in severe fatigue, can induce the ERK MAPK phosphorylation in skeletal
muscle cells (Fitts, 1994). Thus the analogous process of in vivo
myocyte stretching from physical exertion can be seen in myocyte stretching
in vitro.
Ca2+ and positive
feedback hypothesis
The aforementioned response of MAPK to electrical stimulation is noticeable
in fluctuations of established electrochemical gradients within living
cells. Perhaps one of the most salient ions in myocytes is Ca2+. The
presence of calcium ions allow for the operation of the contractile
machinery within myocytes of all types. Via a well-established model,
calcium ions bind to the tropomyosin protein on actin filaments, and
subsequently expose various binding sites for myosin. In this way,
the myosin head can bind to the actin filaments and allow for muscle
contraction to occur. The presence of calcium ions may be correlated
to the presence of mechanical stress on the exterior of the cell since
the influx of ions through stretch sensitive mechanoreceptors has
been implicated in inducing muscle contraction in vitro (Aronson et
al, 2000). Once in the cytosol, Ca2+ ions exert an effect on a variety
of cellular elements. Figure 4 illustrates the indirect role of PKC
in releasing calcium ions. One of the major products of PKC activity,
IP3, is responsible for activating calcium voltage-gated channels
in the membrane of the endoplasmic reticulum. This in vivo process
mimics the activation of mechanoreceptors seen in many in vitro studies
(Aronson et al, 2000). Thus, there appears to be a relationship building
between the role of mechanical cell membrane stretching and calcium
signaling. An intricate network of related pathways seems to be developing,
since it was previously mentioned that PKC activates ERK and p38 pathways.
It is possible to imagine a signaling pathway propagated by calcium
ions that is directly responsible for activating these MAPK pathways.
Such calcium signaling could conceivably originate in a way similar
to the model in figure 5. Such a PKC-mediated pathway, however, has
not yet been experimentally described.
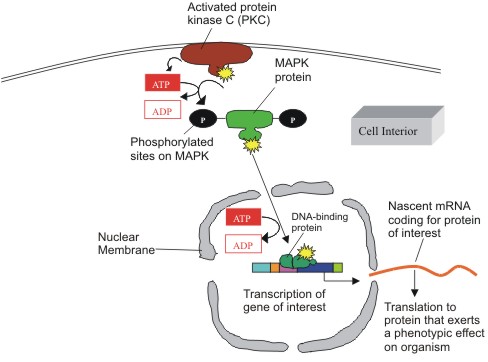 |
Figure
4: Intracellular pathway of MAPK activation via PKC.
Activated PKC phosphorylates MAP kinase on its tyrosine and threonine
sites, at the expense of two ATPs. MAPK then phosphorylates its
downstream targets (not shown) to the level of a transcription
factor that binds to a DNA element and prompts the transcription
of mRNA coding for the protein of interest. Such proteins are
employed in cell differentiation, proliferation, and even death.
(Figure adapted from Alberts B. et al) |
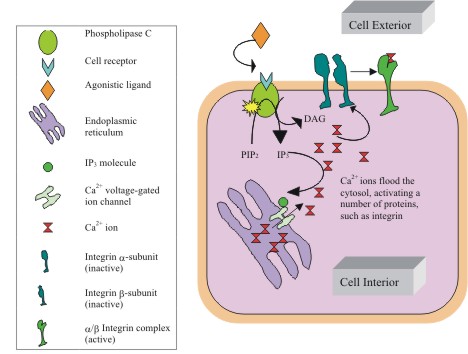 |
Figure
5: Role of IPs in Ca2+ release.
IP3 formation during PKC activation has some interesting effects
within the cytosol. A well-characterized effect of IP3 is binding
to Ca2+ voltage-gated channels, inducing a conformational change
that allows calcium ions to flow down their electrochemical gradient
(from the lumen of the endoplasmic reticulum toward the cell interior).
The presence of calcium ions within the cytosol allows them to
interact with a number of calcium dependent proteins, such as
integrins. Interestingly enough, their binding to such proteins
effectively removes them from the cytosol, thus increasing the
demand for free Ca2+ in the cell interior and stimulating the
release of even more Ca2+ ions. This positive feedback loop will
be important in recruiting multiple myocytes at the tissue level.
(Figure adapted from Voet D., et al) |
A major consideration
not yet addressed is the regulatory mechanism of this postulated signaling
system. In normal physiologic systems, the effects of calcium are
quickly dissipated unless there is a positive feedback mechanism to
fuel the continuation of the calcium-triggered process in question.
The initial release of Ca2+ ions may prompt an even greater release
of calcium into the cytosol, as some ions can bind to allosteric sites
on voltage-gated calcium channels, inducing release from reservoirs
such as the sarcoplasmic reticulum in myocytes (Alberts et al, 1994).
Such a feedback mechanism is supported in evidence of studies on smooth
muscle. These studies illustrated that an initial calcium release
induces an even greater output of systemic calcium to produce an extended
effect, such as prolonged intestinal contraction within the digestive
system (Katoch et al, 1999). Extended contraction is not normally
observed in skeletal muscle, except under tetanic conditions in which
multiple simple muscle spasms are combined into an apparently smooth
continuous effort. Even so, this positive feedback control of calcium
can be involved in systems that sequester calcium for purposes of
signal transduction, as will be discussed next.
INTEGRIN SIGNALING:
A SPECIAL CASE
Integrins comprise a major family of transmembrane proteins that allow
for both cell-cell and cell-matrix associations. Although most integrins
are cell-matrix, those that are involved in cell-cell contacts bind
heterophilically to extracellular matrix elements on adjacent cells.
Integrins are also found in hemidesmosomes (major cell surface attachment
sites at contacts between the cell membrane and components of the extracellular
matrix), where they connect to intermediate filaments inside the cell,
as well as in focal adhesions (cell-matrix adherens junctions) where
they connect to actin filaments and stress fibers (Alberts et al, 1994).
The latter form of integrin interactions proves to be noteworthy in
activating MAPK pathways in response to cell stretching.
There are four major characteristics of integrin that make it particularly
unique as a signaling molecule (Alberts et al, 1994): (1) multiple integrins
each recognize different targets (fibronectins, laminins, etc), (2)
integrins can be regulated (e.g. during mitosis, phosphorylation of
the cytoplasmic tail of the b-subunit of integrin impairs its ability
to bind fibronectin, an extracellular matrix protein involved in cell
adhesion and migration, causing the cells to round up), (3) matrix binding
to integrin regulates cellular activities through focal adhesion kinase
(FAK) (figure 8) signaling cascades, and (4) integrins can be either
Mg2+ or Ca2+ dependent. Each of these four attributes can contribute
to a greater understanding of integrin function at the level of signal
transduction.
Mechanical activation
There are two main subunits for integrin proteins: the a- and the
b-subunit. While there are many binding sites for a variety of proteins
on each of these subunits, the focus will be on the binding site for
laminin, located on the a-subunit, and the binding site for fibronectin
located on the b-subunit (Disatnik et al, 1999). The intracellular
signaling cascades that are activated when integrins bind to their
extracellular ligands are varied. Biochemical changes in cells with
integrin deficiencies indicate that integrins are true signaling molecules,
transmitting information from the extracellular compartment into the
cell in what constitutes "outside-in signaling" (Hynes,
1992). The current fluid-mosaic model of transmembrane proteins in
the plasma membrane and the associated extracellular matrix holds
that the cell membrane components are not static but rather in constant
motion (Alberts et al, 1994). It is not outlandish to consider a force
from mechanical stretching of this fluid membrane that is sufficient
in magnitude to bring together certain transmembrane proteins of interest.
This is in fact what happens with integrins during membrane stretching
(Disatnik et al, 1999). One of the earliest changes initiated by integrin
engagement is clustering of integrins at focal adhesions and tyrosine
phosphorylation of proteins such as paxillin (a cytoskeletal component
that localizes to the focal adhesions at the ends of actin stress
fibers), talin (a cytoplasmic protein that links integrins to the
actin cytoskeleton), and FAK (a cytosolic tyrosine kinase which is
recruited at an early stage to focal adhesions and mediates many downstream
cellular responses) (Schaller et al, 1992). FAK phosphorylation is
considered to be one of the critical steps in the downstream signaling
that promotes cell spreading and cell survival (Disatnik et al, 1999).
Although the details of these more distal events remain to be elucidated,
there is evidence that the binding of integrins to their extracellular
ligands may activate pathways that prevent apoptosis in a variety
of cell types, including skeletal myocytes (Zhang et al, 1995). Association
of integrins with concomitant binding of fibronectin to the b-subunit
is sufficient to form an activated dimer of integrin subunits. Close
inspection of figure 6 illustrates that this aggregation of integrin
leads to the association of various proteins, including FAK and talin,
as well as their localization to focal adhesions (Kornberg et al,
1992).
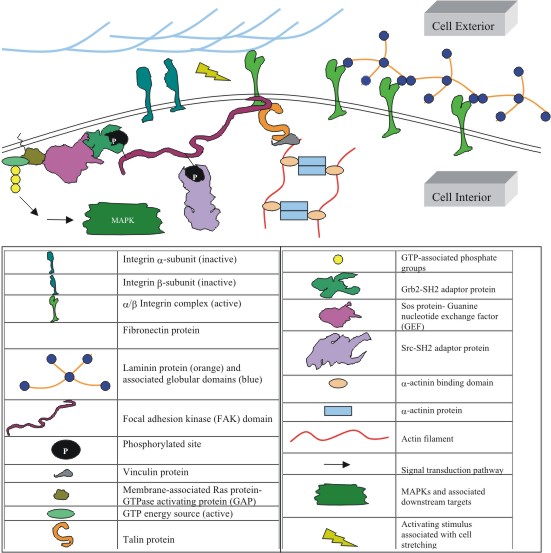 |
Figure
6: Model of MAPK activation via proteins of the extracellular
matrix and cytoskeleton.
Stretching the plasma membrane may cause a number of changes in
the surrounding extracellular matrix and internal cytoskeletal
architecture. These changes may, in turn, activate a MAPK signal
transduction pathway, leading to a number of effects manifested
in cell growth, differentiation, and proliferation. Two mechanisms
may be at work here. First, tension in the cell membrane may prompt
an association of a and b integrin subunits from Ca2+ binding,
thus activating them. These associated units may then further
activate focal adhesion kinase (FAK), with eventual activation
of MAPK. Another possible mechanism may involve the association
of actin filaments to the vinculin linker protein, which may activate
FAK in a similar fashion. The latter mechanism may also involve
the binding of Ca2+, perhaps in preparation of muscle contraction
in active myocytes. In reality, a combination of these two processes
may occur. (Figure adapted from Cooper, et al). |
It has been experimentally
shown that the activation of PKC is necessary for the interaction
of b integrin with fibronectin to promote FAK phosphorylation and
spreading of muscle cells (Disatnik et al, 1999). Woods and Couchman
found that activation of PKC leads to the localization of proteins
such as talin to focal adhesions. Using specific PKC inhibitors, Haimovich
et al. showed that PKC plays a crucial role in integrin signaling
and phosphorylation of FAK in platelets. It has also been shown that
PKC isoforms translocate to nuclear structures and focal adhesions
upon binding of vascular smooth muscle cells to fibronectin (Vuori
et al, 1993). This adds to the growing evidence of the importance
of PKC in both integrin signaling and MAPK activation, which can be
mediated by activated FAK. Since it is known that MAPKs allow for
growth and differentiation of cells (English et al, 1999), the observable
changes in cell spreading induced by associations of integrins, which
indicate growth patterns, must be the result of MAPK signal transduction.
Ionic activation
Integrin subunits also contain binding site for Ca2+ and Mg2+ on both
the a- and b-subunits (Alberts et al, 1994). As noted earlier, one
of the characteristics of integrins is that they are highly regulated,
often by means of these divalent cation binding sites. Integrins can
be activated by Ca2+ binding to its appropriate receptor on the transmembrane
protein. Recalling the previously discussed considerations of calcium
ions in cell stimulus response, it is clear that integrin has an important
role in responding to mechanical stimuli. The interrelationship of
PKC, Ca2+ ions, and integrins is beginning to be revealed: cell stretching
can induce the aggregation of integrins, thus activating them. These
activated integrins may then stimulate the initiation of a signal
cascade through MAPK, in a postulated mechanism delineated in figure
6. The previously discussed Ca2+ positive feedback mechanism mediated
by IP3 binding to the endoplasmic reticulum (figure 3) may constitute
the fuel needed to keep the cycle of MAPK activation running by allowing
more calcium to bind to the integrin divalent cation sites. Furthermore,
the activation of PKC is made possible by the presence of IP3 (figure
3). By syllogism, this is how the action of active PKC is involved
in integrin signaling. This somewhat refines the rather crude model
of the positive feedback mechanism described earlier, in which Ca2+
binds to other voltage gated ion channels, inducing the release of
more calcium ions.
Recruitment of myocytes in tissue systems
The aforementioned considerations have thus far pertained to a single
cell. While it is important to understand the mechanics of cellular
processes, it is equally important to investigate what occurs with
multiple cells at the tissue and organismal level. Taking this into
consideration calls to mind a mechanism of signal spreading, in which
a few cells propagate an activated signal pathway to adjacent, or
nearby cells. Understanding such a mechanism proves to be essential,
since many biochemical processes hardly involve only a single, isolated
cell. Moreover, physiology and pathology are meaningless outside the
context of cell aggregates.
Although integrin engagement leads to signal cascade activation, it
is also clear that the process of muscle cell attachment and spreading
involves an activation of integrins themselves, which then allows
them to execute "inside-out signaling" (Disatnik et al,
1999). In this form of signaling, integrin has an increased affinity
for its extracellular matrix ligand, such as laminin (figures 6 and
7). This activation of integrins by binding laminin promotes the cell
adhesion that may be an important step in the morphological changes
that cells undergo when spreading on a solid substrate (Disatnik et
al, 1999). It turns out that PKC activation is sufficient to promote
inside-out signaling and since it has already been shown that PKC
is necessary for this signaling, a positive feedback loop is created
(Disatnik et al, 1999). Indeed, the gradual morphological changes
associated with cell spreading suggest a multistep process involving
first the detection of the extracellular environment by the cell and
then a progressive change of the cell membrane to interact with that
environment (Disatnik et al, 1999). This is demonstrated most clearly
by the fact that the changes do not occur when cells are plated in
the absence of immobilized matrix proteins to which integrins can
bind (Chen et al, 1994). The presence of such proteins initiates a
signaling cascade inside the cells, and the cells in turn both alter
their membrane properties to interact with the ligands and organize
these ligands into a complex matrix (figure 6). A positive feedback
loop is intrinsic to such a process (Disatnik et al, 1999) and this
further refines the initial feedback model described earlier, thus
completing the understanding of its significance in signal transduction.
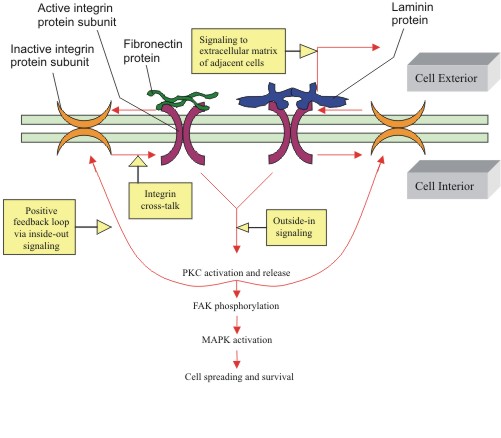 |
Figure
7: Activation of integins via positive feedback sources: outside-in
and inside-out signaling.
Clustering of integrins due to an external stimulus can induce
them to become activated, with subsequent activation of PKC and
downstream phosphorylation of MAPKs. These active integrins can
then activate other neighboring inactive integrins in what constitutes
"integrin cross-talk" via inside-out signaling. This
inside-out signaling proves to be a form of positive feedback,
in which more integrins are recruited to allow for cell spreading
and growth, a phenomenon regulated by MAPKs. Note that since phosphorylation
of FAK is the result of integrin activation, integrins are partially
responsible for initiation of MAPK signal transduction pathways.
(Figure adapted from Disatnik, et al.). |
The positive feedback
loop of integrin engagement, signaling, and activation is shown in
figure 7. Integrins propagate their activated signal to other cells,
via a dynamic equilibrium between an active state and an inactive
state (Disatnik et al, 1999). When there is a sufficient number of
active integrins for effective engagement with their extracellular
ligands, outside-in signaling is initiated, leading to an increase
in PKC activity, a further increase in integrin activation and affinity
(via inside-out signaling), and further outside-in signaling (Disatnik
et al, 1999). This positive feedback loop promotes biochemical changes,
including FAK phosphorylation and focal adhesion formation as seen
in figure 6, which in turn lead to a downstream cascade of biochemical
changes leading to gene expression as shown in figure 4. Inside-out
signaling may also be effective in transducing a signal of interest
to nearby or adjacent cells. The affinity of integrin to laminin,
an extracellular matrix protein involved in cell to cell interactions,
may allow for this to occur. As specific inactive integrins are recruited
to become active, via the proposed positive-feedback mechanism, they
may bind laminins and change conformation. This binding may cause
a widespread agitation in the external environment in a cell, altering
the orientation of the extracellular matrix surrounding neighboring
cells. The fibronectin in these nearby cells may in turn be brought
into closer proximity to the plasma membrane, thus facilitating the
activation of transmembrane proteins, such as integrins, in the same
way as that observed for the initial cell. By this process, integrins
from completely different cells can "talk" to each other
in a rather elaborate communication scheme, allowing for a more global
response to the initiation of a MAPK signal transduction pathway.
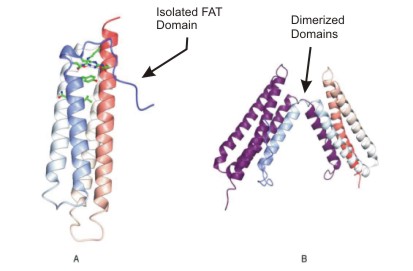 |
Figure
8: Two structures of the focal adhesion targeting domain of focal
adhesion kinase.
The localization of focal adhesion kinase (FAK) to sites of integrin
clustering initiates downstream signaling. The C-terminal focal
adhesion targeting (FAT) domain causes this localization by interacting
with talin and paxillin (not shown). Isolated FAT folds into a
four- helical bundle (A), which has the capacity to form domain-exchanged
dimers in which the N-terminal (B). A structure-based alignment
including these proteins and the vinculin tail domain reveals
a conserved region which could play a role in focal adhesion targeting
(Figure courtesy of Noble, et al). |
CONCLUSION
MAPKs have been shown to be relatively ubiquitious in their activity,
yet their activation is quite specific. This review article has introduced
MAPKs in the conventional light of signal transduction activation: the
binding of a free ligand (i.e. EGF) to its specific receptor, and the
subsequent downstream effects associated with such activation. To provide
a more provocative contrast, the idea of mechanical stretching was introduced
along with its role as a signal transducer. Albeit the binding of tangible
elements from the extracellular matrix is involved in mechanotransduction,
the overall induction of mechanically activated signal transduction
pathways are initially employed by the intangible presence of tension
and mechanical stress in the cell plasma membrane and associated proteins.
To further supplement this unconventional perspective on signal transduction,
the hypothetical model of a positive feedback mechanism was discussed,
involving the employment of calcium ions that originated from the activation
of PKC and other cytosolic components. The final aspect that drove home
the involvement of MAPKs in this feedback mechanism was the observation
of cell spreading and cell growth, two events that are hallmarks of
MAPK activation. While many of the mechanisms and processes discussed
in this review are purely hypothetical, and should be treated as such,
they are the products of the logical synthesis of concepts in MAPK signal
transduction. Thus, in reality, a combination of events such as calcium
activation, rearrangement of extracellular proteins, activation of transmembrane
domains, and transcription at the level of the gene may all be occurring.
Furthermore, these processes may be the result of other components not
discussed in this review. Nonetheless, the models presented in this
article do provide an integrative and innovative approach to signal
transduction that may help in future discoveries.
PROSPECTS
As is the case with most cell analysis, genetic models are always indispensable
in the dissection of various signal transduction pathways. Although
there has been a multitude of such genetic models from which to draw
conclusions as to stress-activated MAPK pathway regulation, new emerging
genetic models such as the dorsal closure pathway in Drosophila, coupled
with the completion of the C. elegans and other genome sequencing projects,
should make it possible to understand the epistatic relationships between
MAPKs and their upstream activators (Tibbles et al, 1999).
Understanding these pathways in the context of human physiology and
disease pathology is much more of a challenge. With regard to skeletal
myocytes in particular, further study of knockout and transgenic mice
may provide the link between signal transduction and muscular dystrophy,
for example. A deficiency of the molecule a7 integrin has been positively
linked to various cases of congenital muscular dystrophy. Muscular dystrophy,
which causes a progressive deterioration of the muscle fiber architecture,
seems to be slightly correlated to the insufficient presence of integrin.
Since it is known that integrin helps establish the complex organization
of the cell membrane, a lack of it must disrupt the integrity of the
membrane leading to the onset of the genetic disease. Thus, a more fundamental
understanding of integrin and related transmembrane proteins may help
assist in elucidating possible treatment, or perhaps even a cure, for
muscular dystrophy. However, paramount discoveries in the exact pathways
of such debilitating muscle disorders can only be realized with a more
aggressive, genetic approach to understanding signal transduction at
its most fundamental core: the activation and initiation of a signaling
pathway. Once this can be fully divulged for a particular skeletal muscle
disease, then perhaps an inhibitory genetic mechanism can be proposed,
in which symptoms can be fully repressed before they are manifested
phenotypically.
At the current state of genetic engineering, the presence of specific
genes in humans cannot be controlled. While this may at first seem like
a major setback for the millions of people afflicted with skeletal muscle
diseases and other genetic ailments alike, there appears to be a great
deal of hope in fighting these diseases using knowledge of signal transduction.
This is to say that while the transmission of a gene from one generation
to another is difficult to control, the first major point of intervention
may be at the level of transcription and translation of a gene, which
is controlled by signal transduction. Gene therapy currently seems to
be a promising solution for treatment of various genetic diseases. Gene
therapy can be targeted to somatic (body) or germ (egg and sperm) cells.
In somatic gene therapy the recipient's genome is changed, but the change
is not passed along to the next generation. In germline gene therapy,
the parents' egg and sperm cells are changed with the goal of passing
on the changes to their offspring. Germline gene therapy is not being
actively investigated, at least in larger animals and humans, although
a lot of discussion is being conducted about its value and desirability.
And while complete obliteration of genetic disease does not seem to
be in the near future, regulating these genetic diseases before they
get out of control is definitely within reach as more research is done
on signal transduction.
ABOUT THE AUTHOR
David A. Barron grew up in El Paso, TX, where he developed an early
affinity for biomedical research through voluntary experiences in the
local area medical center. He attends Rice University, where he is working
towards completion of a B.A. in Biochemistry and Cell Biology. Although
the majority of his coursework is completed at Rice, he has studied
at Oxford University in Oxford, England and through the Baylor College
of Medicine, in the Texas Medical Center, both institutions of which
have enhanced his knowledge of the biosciences tremendously. He is currently
engaged in a research project for the Department of Surgery at Baylor,
examining the mechanical and structural properties of both desmin- and
integrin-deficient mice as seen in biochemical assays and tissue stretching
maneuvers. Past findings have led him into the realm of signal transduction,
specifically investigating the influence of mechanical forces on the
extracellular matrix. The research team he works with, led by Aladin
Boriek, Ph.D., hopes to find a specific role of such mechanical signaling
at the tissue level, with the long term prospect of developing a further
understanding of debilitating muscular diseases resulting from genetic
disorders.
Mr. Barron plans to continue his research during his final year at Rice,
with the possibility of undertaking a joint Rice/Baylor research project
in an attempt reach a synthesis of different ideas in his particular
field of study. While still considering the possibility of pursuing
an M.D./PhD. program, his career goals are primarily oriented towards
earning an M.D. degree in order to apply his knowledge of biochemistry
to patients in a health care setting. In either case, he plans to actively
partake in research projects tailored toward dealing with human disease
at both the macroscopic and microscopic level, so that he may ultimately
examine disease states and pathologic patterns from both a scientific
and clinical perspective.
ACKNOWLEDGEMENTS
Special thanks to Dr. Aladin Boriek and Dr. Ashok Kumar for their patience
and expertise in providing the means for this review. This article would
not have been possible without their guidance and experience in muscle
mechanics and biochemistry. Without a doubt, their words of inspiration
and motivation were the very fuel behind this writing.
FURTHER READING
Burgen A. and Barnard E. A. (1992) Receptor Subunits and Complexes
(Cambridge, England: Cambridge University Press).
Heldin C. H. and Purton M.
(1996) Modular Texts in Molecular and Cell Biology: Signal Transduction,
R. Bradshaw and M. Purton, eds. (New York, New York: Chapman & Hall).
Woodgett R. E. (1994) Frontiers
in Molecular Biology: Protein Kinases (Oxford, England: Oxford University
Press).
REFERENCES
Abe J.I., Kusumara M., Ulevitch
R.J., Berk B.C., and Lee J.D. (1996). Big mitogen-activated protein
kinase 1 (BMK1) is a redox-sensitive kinase. J. Biol. Chem. 271:
16586-16590.
Alberts B., Bray D., Lewis J., Raff M., Roberts K., and Watson J.D.
(1994) Chapter 16: The Cytoskeleton and Chapter 19: Cell Adhesion and
Junctions. Molecular biology of the cell, 3rd edition (New York,
New York: Garland Publishing).
Aronson D, Wojtaszewski J.F., Thorell A., Nygren J., Zangen D., Richter
E.A., Ljungqvist O., Fielding R.A., and Goodyear L.J. (1998). Extracellular-regulated
protein kinase cascades are activated in response to injury in human
skeletal muscle. Am. J. Physiol. 275: C555-561.
Aronson D., Violan M.A., Dufresne S.D., Zangen D., Fielding R.A., and
Goodyear L.J. (1997). Exercise stimulates the mitogen-activated protein
kinase pathway in human skeletal muscle. J. Clin. Invest. 99:
1251-1257.
Aronson D., Wojtaszewski J.F., Thorell A., Nygren J., Zangen D., Richter
E.A., Ljungqvist O., Fielding R.A., and Goodyear L.J. (2000). Differential
activation of mitogen-activated protein kinase signaling pathways by
isometric contractions in isolated slow- and fast-twitch rat skeletal
muscle. Acta Physiol. Scand. 170: 45-49.
Burridge K. and Chrzanowska-Wodnicka M. (1996). Focal adhesions, contractility,
and signaling. Annu. Rev. Cell Dev. Biol. 12: 463-518.
Burridge K., Turner C.E., and Romer L.H. (1992). Tyrosine phosphorylation
of paxillin and pp125FAK accompanies cell adhesion to extracellular
matrix: a role in cytoskeletal assembly. J. Cell Biol. 119:
893-903.
Chao T.H., Hayashi M., Tapping R.I., Kato Y., and Lee J.D. (1999). MEKK3
directly regulates MEK5 as part of the big mitogen-activated protein
kinase 1 (BMK1) signaling pathway. J. Biol. Chem. 274:
36035-36938.
Chen Q., Kinch M.S., Lin T.H., Burridge K., and Juliano R.L. (1994).
Integrin-mediated cell adhesion activates mitogen-activated protein
kinases. J. Biol. Chem. 269: 26602-26605.
Clark E.A. and Brugge J.S. (1995) Integrins and signal transduction
pathways: the road taken. Science 268: 233-269.
Cleland P.J.F., Appleby G.F., Rattigan S., and Clark M.G. (1989). Exercise-induced
translocation of protein kinase C and production of diacylglycerol and
phosphatidic acid in rat skeletal muscle in vivo. Relationship to changes
in glucose transport. J. Biol. Chem. 264: 17704-17711.
Cooper, G.M. (2000) Chapter 13: Cell Signaling. The Cell: A Molecular
Approach 2nd edition (Sunderland, Massachusettes: Washington and
Sinauer Assoc.).
Disatnik M.H. and Rando T.A. (1999) Integrin-mediated muscle cell spreading:
the role of protein kinase C in outside-in and inside-out signaling
and evidence of integrin cross-talk. J. Biol Chem. 274:
32486-32492.
English J., Pearson G., Wilsbacher J., Swantek J., Karandikar M., Xu
S., and Cobb M.H. (1999). New insights into the control of MAP kinase
pathways. Exp. Cell Res. 253: 255-270.
Fitts R.H. (1994). Cellular mechanisms of muscle fatigue. Physiological
Reviews. 74: 49-93.
Gutkind, S.J. (2000). Regulation of mitogen-activated protein kinase
signaling networks by G-protein coupled receptors [online] <http://www.stke.org/cgi/content/full/OC_sigtrans;2000/40/rel>
Haimovich B., Kaneshiki N., and Ji P. (1996). Protein kinase C regulates
tyrosine phosphorylation of pp125FAK in platelets adherent to fibrinogen.
Blood 1: 152-161.
Haller H., Lindschau C., Maash C., Olthoff H., Kurscheid D., and Luft
F.C. (1998). Integrin-induced protein kinase Calpha and Cepsilon translocation
to focal adhesions mediates vascular smooth muscle cell spreading. Circ.
Res. 82: 157-165.
Hanks S.K., Calaib M.B., Harper M.C., and Patel S.K. (1992). Focal adhesion
protein-tyrosine kinase phosphorylated in response to cell attachment
to fibronectin. Proc. Natl. Acad. Sci. U.S.A. 89: 8487-8491.
Hynes R.O. (1992). Integrins: versatility, modulation, and signaling
in cell adhesion. Cell 69: 11-25.
Ilic D., Almeida E.A.C., Schlaepfer D.D., Dazin P., Aizawa S., and Damsky
C.H. (1998). Extracellular matrix survival signals transduced by focal
adhesion kinase suppress p53-mediated apoptosis. J. Cell Biol. 143:
147-160.
Jiang Y., Chen C., Li Z. Guo W., Gegner J.A., Lin S., and Han J. (1996).
Characterization of the structure and function of a new mitogen-activated
protein kinase (p38beta). J. Biol. Chem. 271: 17920-17926.
Juliano R.L. and Haskill S. (1993). Signal transduction from the extracellular
matrix. J. Cell Biol. 120: 577-585.
Katoch S.S., Su X., and Moreland R.S. (1999) Ca2+- and protein kinase
C-dependent stimulation of mitogen-activated protein kinase in detergent-skinned
vascular smooth muscle. Journal of Cellular Physiology 179:
208-217.
Kornberg L., Earp H.S., Parsons J.T., Schaller M., and Juliano R.L.
(1992). Cell adhesion or integrin clustering increases phosphorylation
of a focal adhesion-associated tyrosine kinase. J. Biol. Chem.
267: 23439-23442.
Kyriakis J.M. and Avruch J. (2001). Mammalian Mitogen-Activated Protein
Kinase Signal Transduction Pathways Activated by Stress and Inflammation.
Physiological Reviews 81: 808-869.
Mendelson K.G., Contois L.R., Tevosian S.G., Davis R.J., and Paulson
K.E. (1996). Independent regulation of JNK/p38 mitogen-activated protein
kinases by metabolic oxidative stress in the liver. Proc. Natl. Acad.
Sci. USA 93: 12908-12912.
Noble M.E.M., Ginsberg M., Ladbury J., Werner J., and Campbell I. (2001).
Focal adhesion kinase. Oxford Laboratory of Molecular Biophysics
Online Journal.
Richter E.A., Cleland P. J.F., Rattigan S., and Clark M.G. (1988). Contraction-associated
translocation of protein kinase C in rat skeletal muscle. FEBS Lett.
217: 232-236.
Rozengurt E. and Rodriguez-Fernandéz J.L. (1997). Tyrosine phosphorylation
in the action of neuropeptides and growth factors. Essays Biochem.
32: 73-86.
Ryder J.W., Fahlman R., Wallberg-Henriksson H., Alessi D.R., Krook A.,
and Zierath J.R. (2000). Effect of contraction on mitogen-activated
protein kinase signal transduction in skeletal muscle. Journal of
Biological Chemistry 275: 1457-1462.
Schaller M.D., Borgman C.A., Cobb B.S., Vines R.R., Rynolds A.B., and
Parsons J.T. (1992). pp125FAK a structurally distinctive protein-tyrosine
kinase associated with focal adhesions. Proc. Natl. Acad. Sci. U.S.A.
89: 5192-5196.
Tibbles L.A. and Woodgett J.R. (1999). The stress activated protein
kinase pathways. Cell Mol. Life Sci. 55: 1230-1254.
van Biesen T., Hawes B.E., Raymond J.R., Luttrell L.M., Koch W.A., and
Lefkowitz R.J. (1996). G(o)-protein alpha-subunits activate mitogen-activated
protein kinase via a novel protein kinase C-dependent mechanism. J.
Biol. Chem. 271: 1266-1269.
Voet D. and Voet J.G. (1995) Chapter 17: Glycogen Metabolism. Biochemistry,
2nd edition (New York, New York: John Wiley and Sons, Inc.).
Vuori K. and Ruoslathi E. (1993). Activation of protein kinase C precedes
alpha 5 beta 1 integrin-mediated cell spreading on fibronectin. J.
Biol. Chem. 268: 21459-21462.
Weinberg R. (1998) Chapter 10: Guide proteins of the cell: the machinery
that controls growth. One renegade cell: the quest for the origins of
cancer (Boulder, Colorado: Perseus Books Group).
Widmann C., Gibson S., Jarpe M.B., and Johnson G.L. (1999). Mitogen-activated
protein kinase: conservation of a three-kinase module from yeast to
human. Physiological Reviews 79: 143-180.
Woods A. and Couchman J.R. (1992). Protein kinase C involvement in focal
adhesion formation. J. Cell Sci. 101: 277-290.
Wretman C., Lionikas A., Widegren U., Lannergren J., Westerblad H.,
and Henriksson J. (2001). Effects of concentric and eccentric contractions
on phosphorylation of ERK and p38 MAPKs in isolated rat skeletal muscle.
J. Physiol. 535: 155-164.
Zhang Z., Vuori K., Reed J.C., and Ruoslathi E. (1995). The alpha 5
beta 1 integrin supports survival of cells on fibronectin and up-regulates
Bcl-2 expression. Proc. Natl. Acad. Sci. U.S.A. 92: 6161-6165.
Zhou G, Bao Z.Q., and Dixon J.E. (1995). Components of a new human protein
kinase signal transduction pathway. J. Biol. Chem. 270:
12665-12669.
|
